
Cancer Heterogeneity and Plasticity ISSN 2818-7792
Cancer Heterogeneity and Plasticity 2025;2(2):0011 | https://doi.org/10.47248/chp2502020011
Review Open Access
Microenvironmental Regulation of Colorectal Cancer Stemness: Technological Advances, Molecular Basis, and Therapeutic Targets
Hui Zhao
1,†
 ,
Qilin Li
2,†
,
Jichao Qin
3
,
Qilin Li
2,†
,
Jichao Qin
3
Correspondence: Jichao Qin
Academic Editor(s): Dean G. Tang
Received: Nov 28, 2024 | Accepted: Jun 18, 2025 | Published: Jun 26, 2025
© 2025 by the author(s). This is an Open Access article distributed under the Creative Commons License Attribution 4.0 International (CC BY 4.0) license, which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: Zhao H, Li Q, Qin J. Microenvironmental Regulation of Colorectal Cancer Stemness: Technological Advances, Molecular Basis, and Therapeutic Targets. Cancer Heterog Plast. 2025;2(2):0011. https://doi.org/10.47248/chp2502020011
Stem cell plasticity is crucial for rapidly proliferating tissues, such as the intestine, to cope with microenvironmental stress and maintain homeostasis. This framework may be employed by cancer stem cells (CSCs) to interact with the tumor microenvironment (TME) and give rise to drug resistance, tumor recurrence, and metastasis. Here, we consider colorectal cancer (CRC) as an example to review the technological and conceptual advances in microenvironmental regulation of cancer stemness. Some unique features of CRC, including reprogramming of stromal cells, bacterial niche alteration, and metabolic interplay, were summarized to illustrate how the TME affects the plasticity of CSCs. The key pathways and molecular mechanisms involved in metastasis and drug resistance are reviewed to obtain a deeper understanding of how CSCs may be manipulated for better outcomes in CRC treatment. These findings advance our knowledge of tumor heterogeneity and may provide insights into the development of novel therapeutic strategies.
KeywordsColorectal cancer, tumor microenvironment, cancer stem cells, intestinal stem cells, stem cell plasticity
Colorectal cancer (CRC) is a typical tumor type that illustrates the complexity of cancer stem cells (CSCs) [1,2]. In two pioneering studies, the existence of CSCs was suggested by the finding that self-renewing CRC cells could be enriched using the neural stem cell surface marker CD133 [3,4]. Other cross-tumor CSC markers, such as CD44 and epithelial cell adhesion molecule (EpCAM), have also been used to confirm CSC hierarchy in CRC [5]. Additional evidence comes from the lineage tracking of Lgr5, a marker of murine intestinal stem cells (ISCs). Knockout of Lgr5 not only caused dysfunction of ISCs, but also impaired the self-renewal of CSCs in a mouse model of CRC [6]. As a downstream effector of Lgr5, Wnt signaling is presumed to be the key pathway regulating the self-renewal of ISCs and CSCs in CRC [7]. The literature review establishes a framework for the early CSC concept, which states that the growth of CRC cells is fueled by a small subset of cancer cells with self-renewing and multipotent differentiation capacities (Figure 1); but it does not explain whether the elimination of CSCs alone is sufficient to eradicate CRC. Recently, a study of human organoids indicated that, although the ablation of LGR5+ CSCs led to CRC tumor regression, the spontaneous re-expression of LGR5 in LGR5- cancer cells (i.e., non-CSCs) resulted in tumor regrowth [8]. In addition, our study showed that the transfer of exosomal Wnt from fibroblasts to CD133- cancer cells (non-CSCs) promoted their tumor-initiating capacity and increased the expression of LGR5 in their xenografted progeny [9]. Consistent with the finding that CD133- glioma CSCs also propagated tumors in xenotransplantation assays [10], these results challenged the typical concept of the role of CSCs in solid tumors (Figure 1).

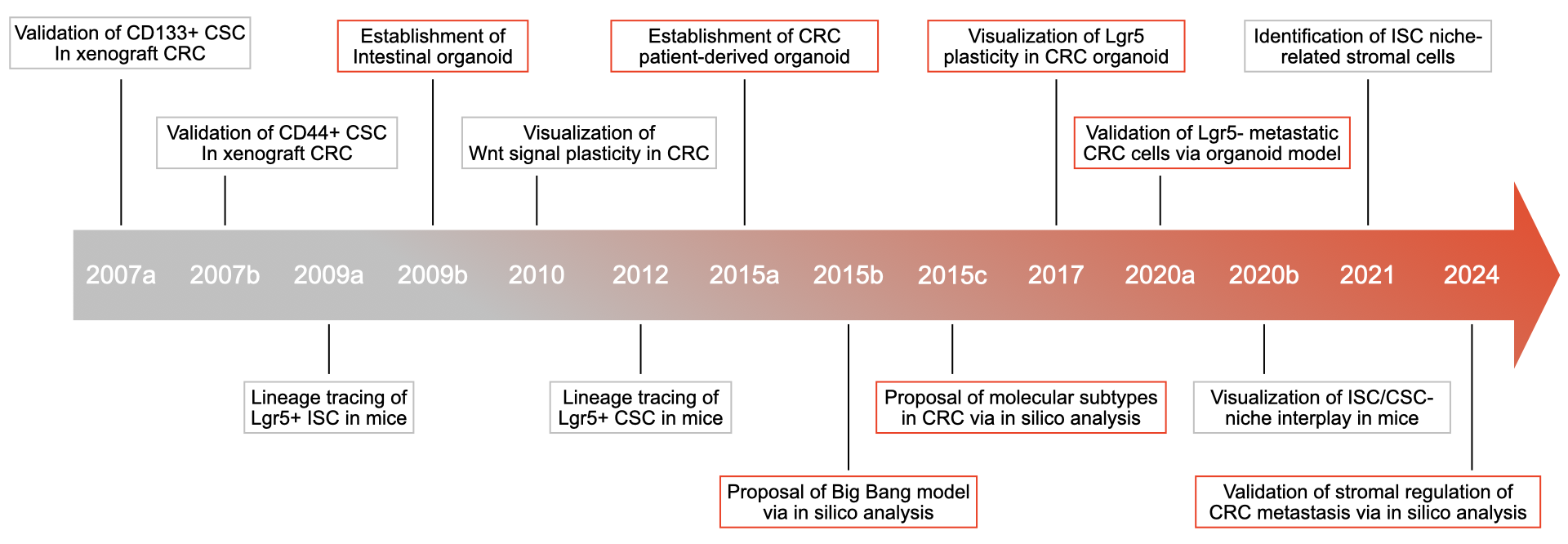
Figure 1 Timeline of the important discoveries clarifying the CSC-niche interplay in CRC.
In this paper, we review the methodologies that were used to investigate the molecular regulation of CSCs [10,11]. Given that CRC is characterized by enhanced CSC-stromal cell crosstalk, significant exposure to the gut microbiome, and the metabolic interdependence between distinct cell populations within the tumor microenvironment, we discuss how these emerging hallmarks affect CSC plasticity in CRC. Furthermore, we summarize the key molecular pathways by which CSCs induce metastasis and drug resistance in patients with CRC.
In the 1990s, Dick et al. employed fluorescence-activated cell sorting (FACS) and xenotransplantation assays to assess the tumorigenic capacity of individual cancer cell subsets in leukemia [12]. This protocol has been widely accepted as the gold standard to evaluate cancer stemness and facilitate the identification of CSC markers in multiple types of tumors, including CRC [10,11]. However, the selection of most cell surface markers is largely dependent on existing knowledge regarding the regulation of stem cells in the brain, breast, and intestines. Anti-CSC strategies based on this data may also impair self-renewal of normal stem cells, particularly those in rapidly proliferating tissues.
Recently, the development of various ‘omics’ and in silico analysis approaches has provided powerful strategies to identify the molecular regulators of CSCs [1]. For instance, Guinney et al. performed an integrated bioinformatics analysis based on 18 existing gene expression datasets from 4151 patients to visualize the molecular landscape of CRC. They identified four consensus molecular subtypes of CRC, including those associated with microsatellite instability, WNT and MYC overexpression, metabolic dysregulation, and mesenchymal properties. The previously mentioned upregulated CSCs markers were placed in the fourth subtype with mesenchymal properties [13].
This study provides global molecular characterization to validate the existence of CSCs in CRC. Similarly, Li et al. performed single-cell RNA sequencing (sc-RNAseq) and spatial transcriptomic analysis of CRC and corresponding liver/ovary metastatic specimens. They identified a stem-like cancer cell cluster, in both primary and metastatic samples, with enhanced expression of the protein tyrosine phosphatase receptor type O (PTPRO) and achaete-like 2 (ASCL2). Notably, the interaction between cancer-associated fibroblasts (CAFs) and endothelial cells via Notch pathways promoted the metastatic capacity of these stem-like CRC cells [14]. In line with these findings, Wang et al. investigated genomic and transcriptomic alterations by matching adjacent normal tissues with primary tumors and metastatic CRC tumors. They described how key mutations in genes of CRC cells, such as KRAS and TP53, modulated PPAR pathways to drive tumorigenesis and metastasis [2]. Although these in silico results need further experimental validation, they expand the study of CSCs and provide a rationale for investigating TME regulation of CSC plasticity.
In 2009, another breakthrough in the CSC field took place when Clevers’ lab reported the establishment of intestinal organoid cultures in vitro using intestinal stem cell (ISC)-dependent three-dimensional (3D) culture technology [15]. These organoids recapitulated the histology and cellular composition of parental intestinal tissues. CRISPR-mediated loss-of-function mutations of APC and TP53 (the most frequent mutation in CRC) in organoids enhanced their ability to grow as CRC tumors in xenotransplantation assays [16]. Subsequently, Clevers’ group generated many patient-derived CRC organoids from clinical samples. Genomics, transcriptomics, epigenomic sequencing, transplantation, and drug screening assays have systematically confirmed that patient-derived organoids (PDOs) can mimic the genotype and phenotype of the parental CRC [17,18]. Given these abilities, PDOs provide novel strategies for lineage-tracing studies. As discovered by Shimokawa et al., LGR5+ CRC cells in PDOs can serially propagate tumors in xenotransplantation assays and differentiate into KRT+ CRC cells. Although genetic ablation of LGR5 protein resulted in CRC regression, unexpectedly a small subset of KRT+ cells may replenish the CSC pool if they spontaneously re-repress LGR5 protein [8]. Moreover, in a liver metastasis model based on orthotopic injection of CRC organoids, genetic ablation of LGR5 rarely reduced tumor growth in the liver. Instead, a proportion of LGR5- CRC cells became highly proliferative and continuously replenished the pool of LGR5+ CSCs [19,20]. Because the maintenance of the ISCs phenotype in LGR5+ cells is regulated by the surrounding stromal niche, it would be interesting to determine if the TME regulates CRC stemness through cancer-stroma crosstalk. In a scRNAseq study based on the co-culture of CRC organoids with corresponding macrophages, overexpression of SPP1 in macrophages promoted CRC development by interacting with CD44 [21]. In other tumors, such as head and neck, breast, and pancreatic cancers, PDOs were further incorporated into CAFs, endothelial cells, neurocytes, and immune cells [22-26]. Undoubtedly, the development of organoid technology will benefit studies on the microenvironmental regulation of CRC stemness (Figure 2).
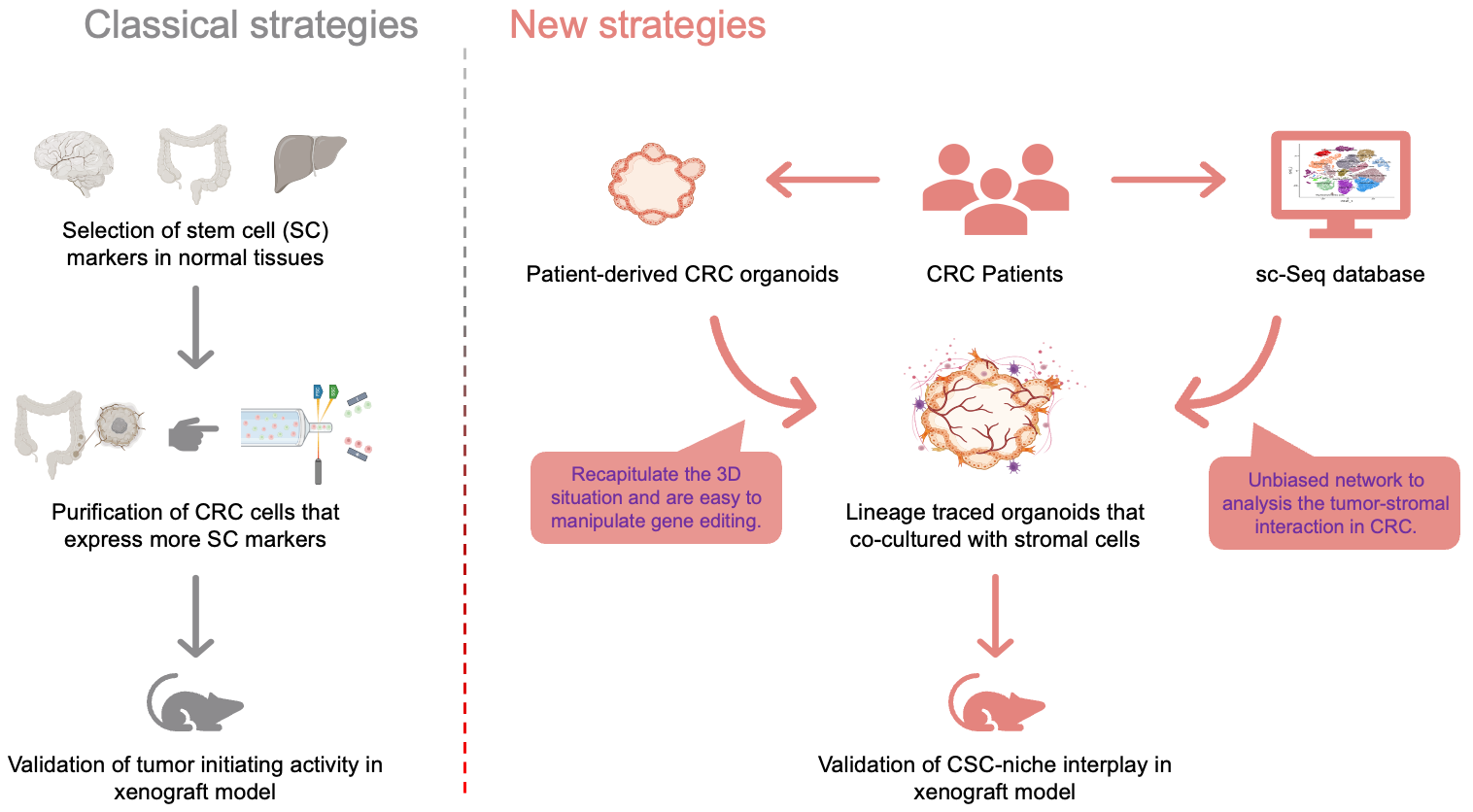
Figure 2 Comparison of classical and new strategies for studying and validating the properties and phenotypic plasticity of CSCs. The development of new technologies, such as single cell sequencing and organoid modeling is expected to advance our knowledge of the role and mechanisms of the influence of CSCs on CRC.
CRC is characterized by an abundance of stromal cells, which counteract microenvironmental stress by secreting cytokines and chemokines to reshape the extracellular matrix (ECM), revascularize the tissue, and harmonize the immune response [27]. Reprogramming of stromal cells is correlated with the formation of a CSC niche. Using scRNA-seq analysis based on the mouse intestine, Roulis et al. found that fibroblasts expressing Ptgs2 (also known as COX-2) constitutively processed arachidonic acid into intermediate prostaglandin H2 (PGH2), which was rapidly converted into highly labile prostaglandin E2 (PGE2) by downstream enzymes such as PTGES. This PGE2 signaling promoted the clonal expansion of Sca-1+ reserve-like stem cells through the Ptger4/Yap cascade. Specific ablation of Ptgs2 in fibroblasts or Ptger4 in Sca-1+ stem cells reduced intestinal regeneration in genetically engineered mice (GEM) and alleviated Apc-induced sporadic tumor initiation in both GEM and PDO models [28]. In another study based on xenotransplantation and 3D co-culture models, secretion of PGE2 by intestinal enteric glia was augmented by tumor cell-derived interleukin-1, and more importantly, promoted the spheroid and xenograft initiating capacity of CD44/CD24+ CRC cells (i.e., CSCs) [29]. Thus, the functional differences in tumorigenic capacities between intestinal stem cells (ISCs) and CRC cells, whose genomes are nearly identical except for the tumor-specific mutations, are primarily governed by divergent gene expression programs shaped through interactions with the surrounding mesenchymal niche (Figure 3).
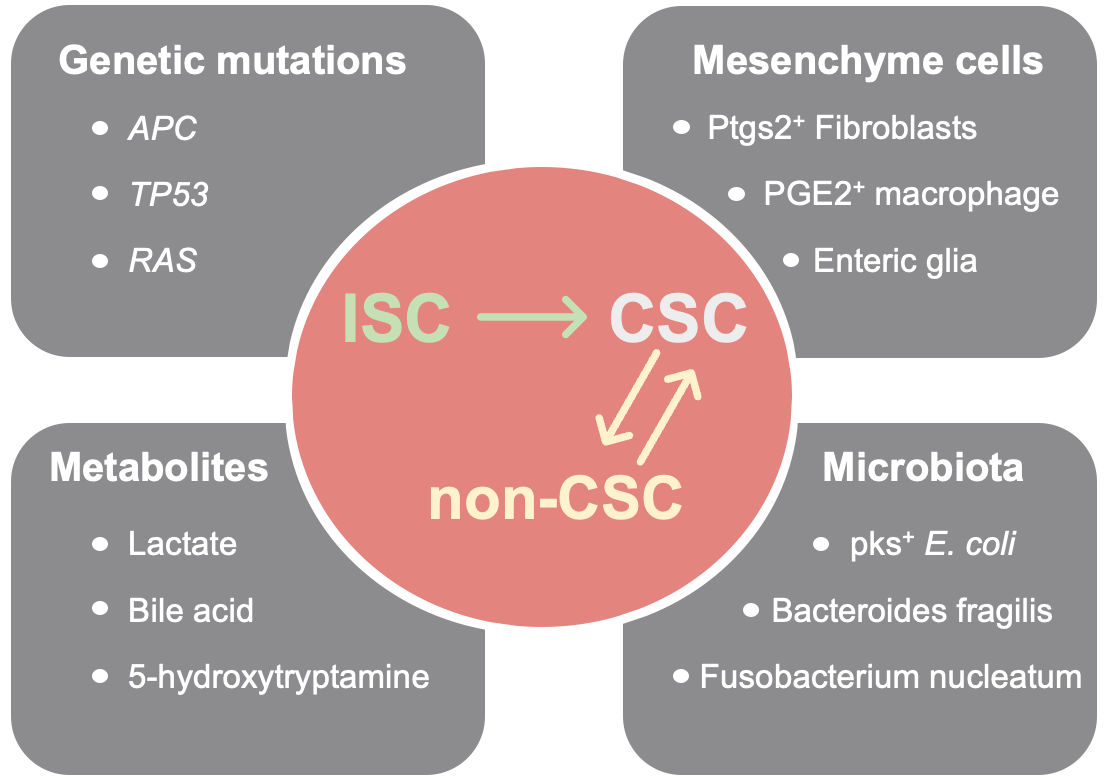
Figure 3 Overview of the emerging hallmarks of tumor microenvironment (TME) that correlate with the regulation of phenotype plasticity of CSCs in CRC.
Intestinal tissues are also specialized by their exposure to a highly dynamic microbiome niche. The ISC-niche interplay can be regulated by reprogramming the gut microbiota. Using GEM and intestinal organoid models, Zhu et al. found that microbiota-derived valeric acid promoted the secretion of 5-hydroxytryptamine (5-HT) in enteric serotonergic neurons to trigger the activation of PGE2+ macrophages, which in turn facilitated Wnt/β-catenin signaling in Lgr5+ ISCs [30]. In their follow-up study of CRC, the connection between 5-HT and the cell surface markers HTR1B/1D/1F suppressed the axin-mediated degradation of β-catenin, and thereby, exacerbated the tumorigenic capacity of CSCs in CRC [31]. The oncogenic properties of Fusobacterium nucleatum (Fn), a typical periodontal disease pathogen, have been thoroughly investigated in recent years. As shown by Zepeda-Rivera et al., the presence of the specific Fn subtype, Fna C2, can be detected in more than 50% of patients with CRC [32]. Zhang et al. employed a genome-wide screening method to determine whether the binding of RadD to CD147 enabled the colonization of Fn in CRC [32]. In addition, binding of Fn-specific FadA to E-cadherin in CRC cells promoted β-catenin signaling [33]. Other intestinal bacteria such as Bacteroides fragilis and pks+ Escherichia coli are also associated with the development of CRC [34]. Investigating the regulation of this niche by the microbiome may improve our understanding of how the intestinal TME modulates CRC stemness.
The metabolic properties of ISCs differ from those of differentiated cells. Specifically, ISCs employ mitochondrial metabolism of glucose and fatty acids to sustain self-renewal, whereas their daughter cells utilize aerobic glycolysis to rapidly fuel their proliferation [35]. The transfer of lactate as a by-product of this metabolic symbiosis can support intestinal tissue homeostasis. Using metabolomic analysis based on an organoid model, Rodríguez-Colman et al. showed that Paneth cell-derived lactate enhanced p38/MAPK signaling in Lgr5+ ISCs by elevating mitochondrial ROS levels. Maintenance of this metabolic interplay contributed to stable intestinal organoid reconstitution [36]. Because ISCs and the CSCs in CRC show similar expression levels of many genes, it is presumed that the same metabolic crosstalk exists in CRC. As a case in point, we compared the metabolic phenotypes of sphere/Wnt+ (CSCs) and adherent/Wnt- cells (non-CSCs) in CRC PDOs. We found that Wnt- cells were proficient in producing lactate to support mitochondrial activity and the ROS/AKT/β-catenin pathway in Wnt+ cells. In transplantation assays with co-implanted spheres and adherent cells, the tumor-initiating activity was reduced when the lactate exporter, MCT4 in non-CSCs, or the transport protein, MCT1 in CSCs was silenced or inhibited [37]. In our follow-up study, the lactate metabolism of CSCs in vivo appeared to be regulated by the oxygen-mediated suppression of HIF-1α and the activation of PGC1-α. As the source of oxygen in the TME, the vasculature in tumors provides a previously overlooked mechanism of regulating CSC phenotypes [38]. Collectively, the metabolic features of individual cell subsets form a complex network that affects CRC stemness (Figure 3).
Dedifferentiation is the process by which differentiated cells are reprogrammed to acquire the ability to self-renew. Investigations of dedifferentiation in CRC have advanced our understanding of stem cell plasticity in solid tumors. Using a WNT reporter system, Vermeulen et al. showed that myofibroblasts secreted growth factors such as HGFs to restore the CSC phenotype in more differentiated CRC cells, both in vitro and in vivo, via a β-catenin-dependent transcriptional network [39]. In line with this, our study showed that secretion of exosomal WNT3A was enhanced in CAFs compared with that in normal fibroblasts, and co-injection of CAFs promoted the tumor-initiating capacity of CD133- CRC cells (non-CSCs) in a xenotransplantation assay [9]. It follows that tumor-initiating capacity is more likely to be described as cancer stemness, which emphasizes it as a flexible state rather than a constant phenotype. Further studies are required to clarify the molecular mechanisms underlying CRC cell dedifferentiation. Hall et al. performed RNAseq analysis in an Apc-deficient mouse model and found that the RNA-splicing factor SRSF1 controlled the plasticity of CRC cells by regulating the splicing level of Kras4b. Moreover, SRSF1 expression affected the reconstitution of CRC PDOs and correlated with the CSCs markers in human CRC samples [40,41]. Hwang et al. isolated colon spheres from primary CRC tissues and profiled their gene expression patterns using microarray analysis. Consequently, the EMT activator, SNAIL, which regulates the expression of IL8 and other genes to sustain cancer stemness in CRC cells [34] was identified. It would be interesting to investigate whether TME signals regulate CRC stemness through these molecular nodes.
The elucidation of normal ISC signaling pathways has made great progress in recent years. However, studies on the abnormal signaling pathways in colorectal CSCs are still in their infancy. Here, we focus on four critical pathways that regulate colorectal CSCs: the Wnt, Hedgehog, BMP (bone morphogenetic protein), and Notch pathways.
Wnt signaling is an essential pathway regulating normal development and physiology and has become adapted to perform various roles in cancer [42,43]. Wnt signal transduction is involved the Wnt/β-catenin pathway, the planar cell polarity (Wnt/PCP) pathway, and the Wnt/Ca2+ signaling pathway [44,45]. Abnormalities in Wnt pathway components have been identified as drivers of cancer and potential targets for treatment, particularly in CRC [17,46]. WNT growth factors are important for the regulation of both normal and malignant stem cells. APC or Wnt/β-catenin mutations exist in 90% of CRCs, increasing Wnt target gene transcription. However, abnormal β-catenin is not sufficient to produce CRC, and other mutations, epigenetic silencing, microenvironmental signals, and cross-pathways help increase nuclear β-catenin to induce tumorigenesis. CRC with Wnt/β-catenin mutations show different levels of Wnt activation, and only high levels of Wnt activation show nuclear localization of β-catenin and possess CSCs characteristics. The Wnt signaling pathway plays a crucial role in CRC cell survival, proliferation, and self-renewal, with dysregulation observed in CSCs [47,48]. ISC dynamics are influenced by several endogenous and exogenous stimuli, including dietary and niche factors. Wnt target genes have the leucine-rich repeat-containing G-protein coupled receptor 5 (Lgr5), the first functional marker of ISCs. Lgr5+ cells, which are an important component of the epithelial stem cell niche, capable of long-term self-renewal and possessing multilineage differentiation potential. The key signaling pathway controlling ISC function is the Wnt/β-catenin pathway, and the Wnt and R-spondin ligands are extensively secreted in the niche, reaching their highest levels at the bottom of the crypt.
Colorectal cancer-related stem cells (CRC-SCs) exhibit distinctive characteristics, including significant heterogeneity and plasticity, contributing to drug resistance and cancer recurrence [49]. When treating patients with CRC, the genetic composition of SCs and their microenvironment aberrantly activate Wnt signaling pathways. CRC-SCs are resistant to both chemotherapy and radiotherapy. LGR5+ CSCs exhibit chemotherapy resistance by entering a quiescent state that allows them to evade drug treatment [50]. The activation of the LRP5 gene further promotes CSC-like phenotypes, including tumorigenicity and resistance to platinum-based drugs in CRC cells, via the canonical Wnt/β-catenin and IL-6/STAT3 signaling pathways [51]. In 2010, Medema et al. identified a CRC that exhibited highly active Wnt signaling in CSCs. This finding suggested that elevated Wnt signaling can be employed as a marker for CRC-SCs, with the Wnt signaling pathway being regulated by the TME [40]. Research has demonstrated a close relationship between Wnt/β-catenin signaling and the ABC transporter of CSCs; also, a critical role of the c-Myc-ABCB5 axis has been implicated in 5-FU resistance in CRC cells. Side population (SP) cells display heightened resistance to 5-FU and irinotecan compared to non-SP cells, in conjunction with the increased activation of the Wnt signaling pathway, inhibition of β-catenin significantly reduced the SP cells [52]. Our study demonstrated that transferring exosomal Wnt from fibroblasts to CD133- cells (non-cancer stem cells) enhanced their tumor-initiating capacity and increased Lgr5 expression in their progeny xenografts [9,53]. Concurrently, we observed that lactate derived from differentiated cells enhanced the Wnt activity of CSCs and promoted their self-renewal in CRC organoids [38,54].
Notch signaling is pivotal in many cellular processes, including stem cell maintenance, cell specification, differentiation, proliferation, and apoptosis [55]. Understanding the intricacies of Notch signaling in its natural context is crucial for uncovering how these pathways may be hijacked in cancer [56]. Numerous studies have elucidated the roles of various Notch components in the regulation of stem cell properties. This raises the question of whether Notch ligand-receptor specificity influences specific subpopulations of stem cells such as CSCs [57]. The heterogeneity of Notch receptors, effectors, and pathways may contribute significantly to CSC diversity [58]. The Notch pathway has been recognized for its modulation of stemness and the tumor-initiating phenotype in CRC; however, results vary depending on the study, model employed, and specific components of the Notch pathway examined [59]. While Notch signaling is often perceived as relatively straightforward, owing to the involvement of only a limited number of proteins, diverse signaling outcomes arise from various combinations of interactions among different ligands and receptors. The role of the Notch pathway in tumors is contingent on the spatial and temporal context of Notch activation and the status of other signaling pathways within cells. This contextual variability likely explains why studies examining the effect of different receptors on CRC stemness have reported a general enhancement of CSC phenotypes despite the presence of non-redundant or even antagonistic effects among the various molecular components involved [60].
A positive correlation has been established between the activation of NOTCH3 and NOTCH1 receptors in CRC. Specifically, NOTCH3 activation occurs by binding the DLL4 ligand, which inhibits the NUMB protein, a known inhibitor of Notch-dependent gene expression. This inhibition of NUMB facilitates the activation of the NOTCH1 receptor, leading to a positive feedback loop that enhances NOTCH3 expression and activation [60]. Notch ligands, particularly JAG1 and DLL4, augment stemness-related characteristics. However, numerous studies investigating the Notch pathway in other pathophysiological contexts have demonstrated distinct or opposing functions of JAG1 and DLL4 ligands, especially regarding their roles in angiogenesis. Both JAG1 and DLL4 ligands are expressed in the embryonic aorta and activate the NOTCH1 receptor, leading to the establishment of hematopoietic and endothelial cell fates, respectively. This ligand-specific lineage differentiation underscores the existence of ligand-receptor specificity within Notch signaling, which is further influenced by crosstalk between Notch ligands [61]. This crosstalk occurs in the intestinal epithelium, where the inhibition of DLL4 or DLL1 does not affect ISCs. However, their simultaneous inactivation disrupts pluripotency and leads to a loss of stem cells (Olfm4+, Lgr5+, and Ascl2+). Nevertheless, the presence of Lgr5+ cells in the intestinal tract of Hes-1, -3, and -5 conditional knockout mice, comparable to that in wild-type mice, indicates that HES transcription factors are not the sole targets of the Notch signaling involved in maintaining pluripotency [62].
Despite numerous studies illustrating the differential effects of the Notch pathway on CRC-SCs, crosstalk among the various components of the Notch pathway and their specific effects complicates the investigation of the effect of these individual proteins on stemness. Few studies have focused on multiple Notch pathway proteins, which are essential for a comprehensive understanding of the influence of the Notch pathway on stemness and self-renewal. These limitations and several other factors must be considered when exploring the relationships among the Notch pathway, stemness phenotypes, CSC heterogeneity, and treatment resistance.
The Hedgehog signaling pathway, which is essential for normal embryonic development, is dysregulated in cancer, leading to tumorigenesis and maintenance of CSCs within the TME [63]. The complex interaction between CSCs and their microenvironment involves various molecular signals that dictate stem cell behavior and fate [64]. Recent studies have highlighted how the Hedgehog pathway influenced CSC functionality by modulating the plasticity in response to microenvironmental cues [65]. For example, specific ligands can activate the Hedgehog signaling cascade, which regulates the expression of genes necessary for maintaining stemness and promoting tumor growth [66]. This signaling directly affects CSCs and alters the surrounding stromal cells, creating a supportive niche that fosters tumor progression and metastasis [67]. Moreover, TME is characterized by a dynamic interplay of various factors, including inflammatory cytokines and the ECM, which can enhance Hedgehog signaling [68]. This interaction can increase the CSC population, contributing to therapeutic resistance and tumor heterogeneity [69]. Understanding these mechanisms is vital for developing targeted therapies to disrupt the CSC niche and improve treatment outcomes for patients with CRC. Zhu et al. reported that TSPAN8 expression was upregulated in breast CSCs, promoted the expression of the stemness genes NANOG, OCT4, and ALDHA1, and was correlated with therapeutic resistance. Mechanistically, TSPAN8 interacts with PTCH1 and inhibits degradation of the SHH/PTCH1 complex by recruiting the de-ubiquitinating enzyme ATXN3 [70]. Gu et al. revealed that circIPO11 drives the self-renewal of liver CSCs and promotes the propagation of HCC (hepatocellular carcinoma) via activation of the Hedgehog signaling pathway. Antisense oligonucleotides (ASOs) against circIPO11 combined with the TOP1 inhibitor camptothecin (CPT) exerted a synergistic antitumor effect [71]. Do et al. discovered that expression of the long non-coding RNA (lncRNA), LOXL1-AS1, was detected in MYCN-expressing Daoy cells and MYCN-amplified SHH-MB (sonic-hedgehog medulloblastomas), and its presence was significantly correlated with reduced survival rates in patients with SHH-MB. Functionally, LOXL1-AS1 enhances cell migration and cancer stemness in SHH-MB cells in vitro. In murine models, MYCN-expressing Daoy cells demonstrated a high metastatic rate and negative effect on survival, both of which were mitigated by perturbation of LOXL1-AS1 [72].
In CRC, abnormal activation of BMP signaling is closely related to tumor progression and the maintenance of stem cell properties [73]. The BMP signaling pathway affects the self-renewal and differentiation abilities of CSCs through transcriptional inhibition mediated by Smad4 [74]. Specifically, BMP signals inhibit transcriptional activity by recruiting the histone deacetylase, HDAC1, to the promoters of stem cell marker genes. In addition, BMP signaling interacts with the Wnt/beta-catenin pathway to regulate CSCs dryness and plasticity of CSCs [75]. Colon CSCs are the main cause of tumor recurrence and drug resistance, and their properties are similar to those of normal stem cells, with the ability to self-renew and undergo multidirectional differentiation [76]. The BMP signaling pathway influences the properties and behavior of CSCs by regulating the stem cell microenvironment and cell-cell interactions. Studies have shown that BMP4 can induce the differentiation of colon CSCs and enhance their responsiveness to chemotherapy [27]. In a mouse model, the administration of BMP4 significantly improved the sensitivity of colon CSCs to chemotherapy drugs, indicating that the BMP signaling pathway has potential applications in regulating the plasticity and therapeutic response of CSCs [77]. Additionally, BMP signaling maintained colon stem cell homeostasis by interacting with Wnt signaling. The Wnt signaling pathway plays an important role in the self-renewal of colon stem cells, and BMP signaling regulates the proliferation and differentiation of stem cells by inhibiting Wnt signaling [78]. This mutually interactive mechanism may be an important regulator of CSCs plasticity in CRC.
In summary, the regulation of CSC plasticity by the TME through the Hedgehog pathway represents a promising area of research with implications for therapeutic strategies targeting CSCs and the TME.
The primary distinction between stem cells and other cell types lies in their epigenetic architecture [79,80]. Epigenetic regulators, transcription factors, and microRNAs (miRNAs) are crucial for maintaining stem cell self-renewal. The pervasive aberrant epigenetic regulation observed in tumors indicates a strong association between epigenetic mechanisms and cancer [81]. Advances in sequencing technologies have significantly enhanced the detection of rare yet functionally significant epigenetic events, thereby contributing substantially to the precise targeting of cancer cells [82]. Epigenetic regulatory mechanisms play a pivotal role in the plasticity of CSCs, and the TME has a significant influence [83]. The interplay between epigenetic modifications and the microenvironment can determine the fate of CSCs, affecting their self-renewal capacity, differentiation potential, and contribution to tumor heterogeneity [84]. Wang et al. reported that BEX1 played a key role in regulating CSCs properties in different types of liver cancer. Targeting BEX1-mediated Wnt/β-catenin signaling may help address the high recurrence rate and heterogeneity of liver cancer [85]. The TME comprises diverse cellular and non-cellular elements encompassing the ECM, stromal cells, and immune cells, all of which provide biochemical and mechanical cues that shape the epigenetic profile of CSCs. For instance, changes in ECM rigidity and composition can alter the chromatin structure and gene expression, thereby promoting a more adaptable state in cancer cells. This adaptability enables CSCs to respond to the dynamic milieu of the tumor microenvironment, facilitating processes such as metastasis and therapy resistance.
DNA methylation is one of the most prevalent epigenetic modifications and involves DNA methyltransferases (DNMTs) which add methyl groups to the fifth carbon atom of the cytosine ring to form 5-methylcytosine (5mC) [86,87]. In normal cells, DNA methylation primarily occurs in CpG dinucleotide sequences on CpG islands and plays a role in gene silencing and preserving genomic stability [88,89]. In contrast, tumors simultaneously exhibit global hypomethylation and hypermethylation of CpG islands, resulting in altered gene expression profiles that can foster cancer initiation and progression [90,91]. Within the TME, stress signals such as hypoxia, inflammation, and cytokine release can trigger the upregulation of DNMT expression, leading to hypermethylation of the promoter regions of crucial stemness-related genes such as Oct4, Sox2, and Nanog [92], which inhibits the self-renewal capacity of the CSCs. Conversely, the TME can lower the overall genome methylation level by diminishing the activity of DNMTs or activating demethylases, thereby reactivating previously silenced oncogenes and potentially enhancing the aggressiveness and resistance of CSCs to chemotherapy.
Histone modification is a crucial epigenetic regulatory mechanism encompassing acetylation, methylation, ubiquitination, phosphorylation, and SUMOylation [77,93]. These modifications affect DNA accessibility and binding efficiency of transcription factors by reshaping the chromatin structure, thereby orchestrating gene expression [85,87]. Histone acetylation is typically associated with an open chromatin state that promotes active gene expression [94]. At the same time, the effects of methylation with its complex regulation, is contingent upon the specific histone residues altered and the extent of modification [95]. Within the TME, various enzymes involved in histone modification, such as histone deacetylases (HDACs), histone demethylases, lysine-specific methyltransferases (KMTs) and demethylases undergo modulation, thereby reshaping the expression profile of stem cell-related genes in CSCs. Bi et al. found that HDAC11 suppressed LKB1 expression in HCC to promote cancer stemness, progression, and sorafenib resistance, suggesting the potential of targeting HDAC11 to treat HCC and overcome kinase inhibitor resistance [96]. Notably, HDAC inhibitors have been shown to reverse the stemness of CSCs, diminish the efficiency of tumor spheroid formation, and increase their sensitivity to chemotherapeutic agents. Caslini et al. proposed that the inactivation of HDAC7, either directly or via the inhibition of HDAC1 and HDAC3, may lead to the suppression of the CSCs phenotype by downregulating several super-enhancer-associated oncogenes. The selective targeting of CSCs by this mechanism and the potential to concurrently inhibit multiple oncogenes underscores the importance of developing HDAC7-specific inhibitors as promising research objectives [97].
Owing to their distinctive roles in gene expression regulation, regulatory networks mediated by noncoding RNAs have gained much attention recently, particularly miRNAs and lncRNAs [98]. MicroRNAs function by negatively regulating gene expression through complementary base pairing with the 3’UTR of the target mRNA, leading to either translation inhibition or mRNA degradation [99]. In contrast, lncRNAs act as competitive endogenous RNAs (sponges) for miRNAs or interact with transcription factors, RNA polymerase II (Pol II), and other epigenetic regulators to positively or negatively modulate the expression of target genes [100]. In the TME, factors such as hypoxia, inflammatory mediators, and intercellular communication can alter the expression levels and activities of non-coding RNAs, consequently affecting the proliferation, migration, chemotherapeutic resistance, and other functions of CSCs [101]. For example, several miRNAs, including miR-145, miR-34a, and members of the let-7 family, are downregulated in various types of cancer. Overexpression of these miRNAs suppresses the expression of CSC markers, reduces the formation of tumor spheroids, and inhibits tumorigenesis and chemotherapy resistance in vivo. The lncRNAs, MALAT1, HOTAIR, and H19, have been implicated in CSC regulation within the TME [102]. These lncRNAs act as miRNA sponges or cooperate with histone-modifying enzymes to regulate downstream target gene expression, thereby influencing the fate and functional traits of CSCs.
In addition to the epigenetic mechanisms discussed earlier, the role of chromatin remodeling complexes in CSC plasticity is crucial [103]. These complexes consist of a set of proteins that manipulate the position of nucleosomes via ATP hydrolysis [104]. They either expose or conceal transcription initiation sites in DNA, consequently regulating gene expression [105,106]. Within the TME, factors such as hypoxia, cytokines, and ECM components modulate the localization of CSCs, consequently affecting the chromatin accessibility and activity of crucial genes [107]. For instance, Portney et al. demonstrated that a mere two days of transient induction of ZSCAN4 (zinc finger and SCAN domain-containing 4) resulted in an increased frequency of CSCs both in vitro and in vivo, accompanied by the upregulation of pluripotency and CSC-associated factors. Notably, we elucidated, for the first time, the role of ZSCAN4 in modulating the epigenetic landscape and regulating chromatin configuration. Our findings indicated that ZSCAN4 induced functional hyperacetylation of histone three at the OCT3/4 and NANOG promoters, thereby facilitating the upregulation of CSC factors [108]. The SWI/SNF family members are important chromatin remodeling factors capable of opening densely packed heterochromatin regions, facilitating the access of transcription factors to DNA, thereby promoting the transcription of downstream genes related to plasticity [109]. However, in certain instances, the SWI/SNF complex participates in heterochromatin assembly and suppresses the expression of specific genes. Members of the ISWI family can also alter the TME by compacting chromatin, repressing gene transcription, and restricting the maintenance, proliferation, and differentiation of CSCs (Figure 4).
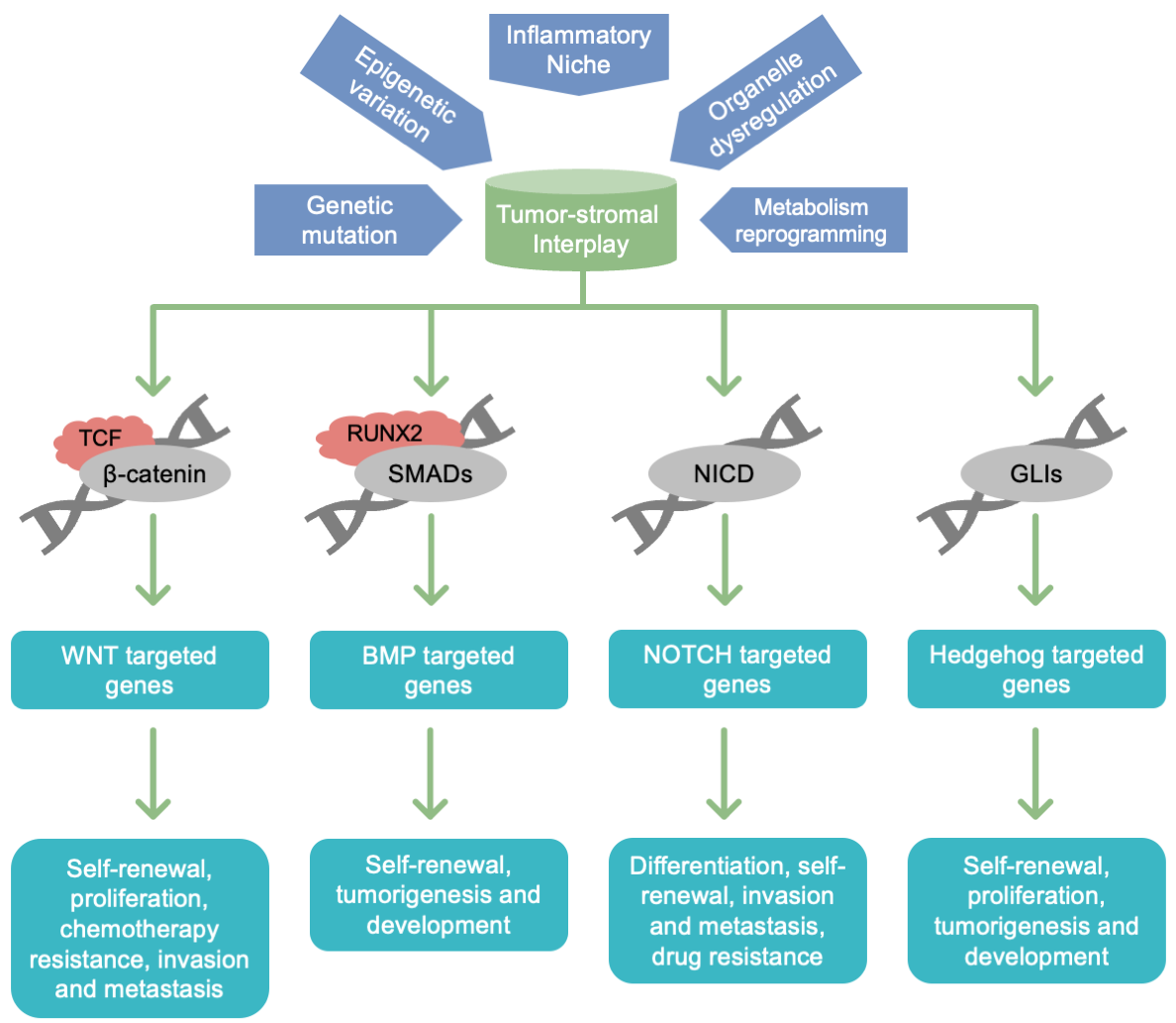
Figure 4 Key functional pathways of CSC action as TME pivots during the course of tumor progression in patients with CRC.
Metastasis is a major cause of cancer-related death worldwide. Several studies have reported the correlation between CSCs and CRC metastasis. Using spatial transcriptomics (ST) technology, Zhou et al. compared the tissue-wide transcriptome information of primary CRC with that of corresponding metastatic liver specimens. The expression of CSC markers is enriched in metastatic tissues, and FOXD1 was selected as a marker to predict metastatic potential [110]. In a xenotransplantation model, overexpression of protease-activated receptor 2 (PAR2) enhanced the self-renewal and metastatic capacity of CSCs by triggering β-catenin-mediated transcription of periostin, a metastasis-related ECM protein [111]. Similarly, Wang et al. showed that PGE2 induced CSCs expansion by activating the EP4-PI3K/MAPK/NF-κB signaling axis and promoted the formation of liver metastases in GEM and orthotopic transplantation models [112]. These studies provided clues for targeting CSCs to prevent metastasis. But whether targeting CSCs alone is sufficient to block metastasis remains unclear. As shown by Gavert et al., IL-1 enhanced CRC cell motility and invasion, and promoted liver metastasis in vivo. This process is rarely dependent on changes in CSCs markers such as CD133, CD44, and EpCAM [113]. Furthermore, de Sousa and Melo et al. validated the distinct role of Lgr5+ CSCs in primary and metastatic CRC in a lineage-tracing GEM model. In liver metastases, where the TME is largely different from that in primary CRC, the ablation of Lgr5+ CSCs rarely affected tumor growth [19,114]. Metastasis requires cancer cells to invade the surrounding niche, disseminate into the circulation, enter and exit quiescence, and colonize distant organs [115]. A better explanation is that CSCs are more proficient in counteracting the TME to survive the metastatic cascade. Todaro et al. showed that overexpression of CD44v6 in CSCs enhanced their ability to drive CRC metastasis through their capacity for migration [116]. In another study, Li et al. showed that overexpression of PTPRO and ASCL2 facilitated the interaction between CSCs, CAFs, and endothelial cells to promote metastasis [117]. In another study on breast cancer, PGC-1α-mediated mitochondrial biogenesis enhanced the survival of circulating tumor cells (CTCs) [55]. This is consistent with our finding that overexpression of PGC-1α in CRC-CSCs promoted their metastatic capacity in vivo [39]. Thus, a deeper understanding of how CSCs survive the metastatic cascade may benefit the development of anti-metastatic strategies.
CSCs are well known for their ability to give rise to resistance against chemotherapy, radiotherapy, and immune and targeted therapies [118]. A typical explanation is the inherent overexpression of ATP-binding cassette (ABC) transporter family proteins in CSCs [119]. In CRC, the expression of the ABC transporter appears to be regulated by SOX2 and PGC1-α [120,121]. Targeting ABC protein-related signals impairs the efflux of anticancer drugs driven by CSCs [121]. Because of their specific transcription networks, CSCs may also develop other drug-resistance mechanisms. Mangiapane et al. showed that PI3K-driven overexpression of human epidermal growth factor receptor 2 (HER2) sustained the interaction of CSCs with cancer-associated fibroblasts to promote CRC resistance to anti-EGFR therapy. The blockade of this signal reduced the dissemination of CSCs in adjuvant therapy [122]. Yuan et al. showed that the CSC marker, CD133, directly interacted with ABCC1 protein through the AKT/NF-κB signaling axis to augment chemotherapy resistance in CRC cells, providing a rationale for targeting the cooperation between two individual CSC-related molecules [123].
The use of scRNAseq technology has advanced our understanding of how the TME regulates therapy resistance. Li et al. employed scRNAseq to investigate the dynamics of immune and stromal cells in 19 patients with CRC receiving immune checkpoint inhibitor (ICI) therapy. They found that CRC resistance to PD-1 blockade was associated with the absence of CCL2+ fibroblasts and the presence of HLA-DRA+ endothelial cells [124]. It would be worthwhile investigating how TME affects therapeutic resistance by regulating the interactions of CSCs with these stromal cell populations.
Acquired drug resistance is another mechanism by which CSCs cause failure of clinical treatment. Using a panel of CRC PDOs, Solé et al. found that DNA-damaging agents induced a persistent quiescent-like phenotype by triggering the YAP-dependent acquisition of the fetal stem cell signature in P53 wild-type CRC cells [125]. These findings highlight the correlation between stem cell plasticity and chemotherapy resistance in patients with CRC. Consistent with this, Dong et al. established several 5-FU-resistant CRC cell lines and showed that HIF-1α-mediated glucose metabolism reprogramming promoted the survival of CRC cells via activating the Wnt/β-catenin signal [126]. Using an in silico model, Woolston et al. analyzed the genomic and transcriptomic landscapes of patients with cetuximab-resistant CRC. Consequently, most biopsies with acquired resistance harbored no genetic resistance drivers; instead, interactions between fibroblasts and growth factors aggravated drug resistance in CRC cells [127]. (Table 1)
Table 1. Potential targets by which CSCs lead to CRC progression.
Overall, the hypothesis that the CSC phenotype is plastic has been widely accepted and thoroughly validated in CRC. Self-renewal and differentiation of CSCs, as well as dedifferentiation of non-CSCs appears to be regulated by a series of factors, including key driver mutations of CRC, the inflammatory niche stromal cells, specific metabolites and intestinal microbiota (Figure 3). Development of multi-omics sequencing (omics approaches, including proteomics or epigenomics) and in silico analyses facilitated investigation of the above mechanisms, and elucidated the indispensable role of the TME in regulating the plasticity of CSCs. Construction of human organoid models provides a novel strategy to identify the key drivers of mutated ISCs, the central pathways of CSC plasticity, and the interplay of CSCs with the surrounding niche. Of note, TME regulation of CSCs in CRC may be further complicated by frequent exposure to chyme, metabolites, and bacteria, but how these exogenous factors affect CSC properties remains unclear. In addition, the targeting of CSCs in CRC still faces considerable challenges due to the lack of specific markers. In future, a combination of biotechnology, bioinformatics, and artificial intelligence technology should provide keen insights into the intimate relationships between CSCs and CRC, hopefully providing a new group of targets for therapeutic exploitation.
Not applicable.
Not applicable.
The authors declare that no competing interests exist.
This study was supported by grants from the National Natural Science Foundation of China (No. 82173368 and 82372995 to J.Q.).
The authors utilized Pivot science publication editorial services to revise the manuscript and did not employ any ChatGPT model to improve the readability and language of this work.
| 1. | Frank MH, Wilson BJ, Gold JS, Frank NY. Clinical Implications of Colorectal Cancer Stem Cells in the Age of Single-Cell Omics and Targeted Therapies. Gastroenterology. 2021;160(6):1947-1960. [Google Scholar] [CrossRef] |
| 2. | Lin K, Chowdhury S, Zeineddine MA, Zeineddine FA, Hornstein NJ, Villarreal OE, et al. Identification of Colorectal Cancer Cell Stemness from Single-Cell RNA Sequencing. Mol Cancer Res. 2024;22(4):337-346. [Google Scholar] [CrossRef] |
| 3. | O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106-110. [Google Scholar] [CrossRef] |
| 4. | Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445(7123):111-115. [Google Scholar] [CrossRef] |
| 5. | Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007;104(24):10158-10163. [Google Scholar] [CrossRef] |
| 6. | Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457(7229):608-611. [Google Scholar] [CrossRef] |
| 7. | Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337(6095):730-735. [Google Scholar] [CrossRef] |
| 8. | Shimokawa M, Ohta Y, Nishikori S, Matano M, Takano A, Fujii M, et al. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature. 2017;545(7653):187-192. [Google Scholar] [CrossRef] |
| 9. | Hu YB, Yan C, Mu L, Mi YL, Zhao H, Hu H, et al. Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene. 2019;38(11):1951-1965. [Google Scholar] [CrossRef] |
| 10. | Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23(10):1124-1134. [Google Scholar] [CrossRef] |
| 11. | Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21(3):283-296. [Google Scholar] [CrossRef] |
| 12. | Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730-737. [Google Scholar] [CrossRef] |
| 13. | Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350-1356. [Google Scholar] [CrossRef] |
| 14. | Wang R, Li J, Zhou X, Mao Y, Wang W, Gao S, et al. Single-cell genomic and transcriptomic landscapes of primary and metastatic colorectal cancer tumors. Genome Med. 2022;14(1):93. [Google Scholar] [CrossRef] |
| 15. | Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;521(7550):43-47. [Google Scholar] [CrossRef] |
| 16. | van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161(4):933-945. [Google Scholar] [CrossRef] |
| 17. | de Sousa EM, Vermeulen L, Richel D, Medema JP. Targeting Wnt signaling in colon cancer stem cells. Clin Cancer Res. 2011;17(4):647-653. [Google Scholar] [CrossRef] |
| 18. | Fujii M, Shimokawa M, Date S, Takano A, Matano M, Nanki K, et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell. 2016;18(6):827-838. [Google Scholar] [CrossRef] |
| 19. | Fumagalli A, Oost KC, Kester L, Morgner J, Bornes L, Bruens L, et al. Plasticity of Lgr5-Negative Cancer Cells Drives Metastasis in Colorectal Cancer. Cell Stem Cell. 2020;26(4):569-578.e567. [Google Scholar] [CrossRef] |
| 20. | Li N, Zhu Q, Tian Y, Ahn KJ, Wang X, Cramer Z, et al. Mapping and modeling human colorectal carcinoma interactions with the tumor microenvironment. Nat Commun. 2023;14(1):7915. [Google Scholar] [CrossRef] |
| 21. | LeSavage BL, Suhar RA, Broguiere N, Lutolf MP, Heilshorn SC. Next-generation cancer organoids. Nat Mater. 2022;21(2):143-159. [Google Scholar] [CrossRef] |
| 22. | Holkom M, Yang X, Li R, Chen Y, Zhao H, Shang Z. Fibroblast regulates angiogenesis in assembled oral cancer organoid: A possible role of NNMT. Oral Dis. 2024;30(8):4982-4992. [Google Scholar] [CrossRef] |
| 23. | Zhao H, Jiang E, Shang Z. 3D Co-culture of Cancer-Associated Fibroblast with Oral Cancer Organoids. J Dent Res. 2021;100(2):201-208. [Google Scholar] [CrossRef] |
| 24. | Zhao H, Li R, Chen Y, Yang X, Shang Z. Stromal nicotinamide N-methyltransferase orchestrates the crosstalk between fibroblasts and tumour cells in oral squamous cell carcinoma: evidence from patient-derived assembled organoids. Oncogene. 2023;42(15):1166-1180. [Google Scholar] [CrossRef] |
| 25. | Tuveson D, Clevers H. Cancer modeling meets human organoid technology. Science. 2019;364(6444):952-955. [Google Scholar] [CrossRef] |
| 26. | Wu N, Sun H, Zhao X, Zhang Y, Tan J, Qi Y, et al. MAP3K2-regulated intestinal stromal cells define a distinct stem cell niche. Nature. 2021;592(7855):606-610. [Google Scholar] [CrossRef] |
| 27. | Medema JP, Vermeulen L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature. 2011;474(7351):318-326. [Google Scholar] [CrossRef] |
| 28. | Roulis M, Kaklamanos A, Schernthanner M, Bielecki P, Zhao J, Kaffe E, et al. Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature. 2020;580(7804):524-529. [Google Scholar] [CrossRef] |
| 29. | Vales S, Bacola G, Biraud M, Touvron M, Bessard A, Geraldo F, et al. Tumor cells hijack enteric glia to activate colon cancer stem cells and stimulate tumorigenesis. EBioMedicine. 2019;49:172-188. [Google Scholar] [CrossRef] |
| 30. | Zhu P, Lu T, Wu J, Fan D, Liu B, Zhu X, et al. Gut microbiota drives macrophage-dependent self-renewal of intestinal stem cells via niche enteric serotonergic neurons. Cell Res. 2022;32(6):555-569. [Google Scholar] [CrossRef] |
| 31. | Zhu P, Lu T, Chen Z, Liu B, Fan D, Li C, et al. 5-hydroxytryptamine produced by enteric serotonergic neurons initiates colorectal cancer stem cell self-renewal and tumorigenesis. Neuron. 2022;110(14):2268-2282.e4. [Google Scholar] [CrossRef] |
| 32. | Zepeda-Rivera M, Minot SS, Bouzek H, Wu H, Blanco-Miguez A, Manghi P, et al. A distinct Fusobacterium nucleatum clade dominates the colorectal cancer niche. Nature. 2024;628(8007):424-432. [Google Scholar] [CrossRef] |
| 33. | Zhang L, Leng XX, Qi J, Wang N, Han JX, Tao ZH, et al. The adhesin RadD enhances Fusobacterium nucleatum tumour colonization and colorectal carcinogenesis. Nat Microbiol. 2024;9(9):2292-2307. [Google Scholar] [CrossRef] |
| 34. | Hwang WL, Yang MH, Tsai ML, Lan HY, Su SH, Chang SC, et al. SNAIL regulates interleukin-8 expression, stem cell–like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology. 2011;141(1):279-291.e1-5. [Google Scholar] [CrossRef] |
| 35. | Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14(2):195-206. [Google Scholar] [CrossRef] |
| 36. | Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029-1033. [Google Scholar] [CrossRef] |
| 37. | Rodriguez-Colman MJ, Schewe M, Meerlo M, Stigter E, Gerrits J, Pras-Raves M, et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature. 2017;543(7645):424-427. [Google Scholar] [CrossRef] |
| 38. | Zhao H, Yan C, Hu Y, Mu L, Liu S, Huang K, et al. Differentiated cancer cell-originated lactate promotes the self-renewal of cancer stem cells in patient-derived colorectal cancer organoids. Cancer Lett. 2020;493:236-244. [Google Scholar] [CrossRef] |
| 39. | Liu S, Zhao H, Hu Y, Yan C, Mi Y, Li X, et al. Lactate promotes metastasis of normoxic colorectal cancer stem cells through PGC-1alpha-mediated oxidative phosphorylation. Cell Death Dis. 2022;13(7):651. [Google Scholar] [CrossRef] |
| 40. | Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12(5):468-476. [Google Scholar] [CrossRef] |
| 41. | Hall AE, Pohl SO, Cammareri P, Aitken S, Younger NT, Raponi M, et al. RNA splicing is a key mediator of tumour cell plasticity and a therapeutic vulnerability in colorectal cancer. Nat Commun. 2022;13(1):2791. [Google Scholar] [CrossRef] |
| 42. | Shenoy AK, Fisher RC, Butterworth EA, Pi L, Chang LJ, Appelman HD, et al. Transition from colitis to cancer: high Wnt activity sustains the tumor-initiating potential of colon cancer stem cell precursors. Cancer Res. 2012;72(19):5091-5100. [Google Scholar] [CrossRef] |
| 43. | Agudo J, Miao Y. Stemness in solid malignancies: coping with immune attack. Nat Rev Cancer. 2024;25:27-40. [Google Scholar] [CrossRef] |
| 44. | Nik AM, Reyahi A, Ponten F, Carlsson P. Foxf2 in intestinal fibroblasts reduces numbers of Lgr5(+) stem cells and adenoma formation by inhibiting Wnt signaling. Gastroenterology. 2013;144(5):1001-1011. [Google Scholar] [CrossRef] |
| 45. | Sarabia-Sanchez MA, Moreno-Londono AP, Castaneda-Patlan MC, Alvarado-Ortiz E, Martinez-Morales JC, Robles-Flores M. Non-canonical Wnt/Ca2+ signaling is essential to promote self-renewal and proliferation in colon cancer stem cells. Front Oncol. 2023;13:1121787. [Google Scholar] [CrossRef] |
| 46. | Shiokawa D, Sakai H, Ohata H, Miyazaki T, Kanda Y, Sekine S, et al. Slow-Cycling Cancer Stem Cells Regulate Progression and Chemoresistance in Colon Cancer. Cancer Res. 2020;80(20):4451-4464. [Google Scholar] [CrossRef] |
| 47. | Hua F, Shang S, Yang YW, Zhang HZ, Xu TL, Yu JJ, et al. TRIB3 Interacts With beta-Catenin and TCF4 to Increase Stem Cell Features of Colorectal Cancer Stem Cells and Tumorigenesis. Gastroenterology. 2019;156(3):708-721.e15. [Google Scholar] [CrossRef] |
| 48. | Herpers B, Eppink B, James MI, Cortina C, Canellas-Socias A, Boj SF, et al. Functional patient-derived organoid screenings identify MCLA-158 as a therapeutic EGFR x LGR5 bispecific antibody with efficacy in epithelial tumors. Nat Cancer. 2022;3(4):418-436. [Google Scholar] [CrossRef] |
| 49. | Tang Q, Chen J, Di Z, Yuan W, Zhou Z, Liu Z, et al. TM4SF1 promotes EMT and cancer stemness via the Wnt/beta-catenin/SOX2 pathway in colorectal cancer. J Exp Clin Cancer Res. 2020;39(1):232. [Google Scholar] [CrossRef] |
| 50. | Park SY, Kim JY, Jang GB, Choi JH, Kim JH, Lee CJ, et al. Aberrant activation of the CD45-Wnt signaling axis promotes stemness and therapy resistance in colorectal cancer cells. Theranostics. 2021;11(18):8755-8770. [Google Scholar] [CrossRef] |
| 51. | Nie X, Liu H, Ye W, Wei X, Fan L, Ma H, et al. LRP5 promotes cancer stem cell traits and chemoresistance in colorectal cancer. J Cell Mol Med. 2022;26(4):1095-1112. [Google Scholar] [CrossRef] |
| 52. | Zhu Y, Li X. Advances of Wnt Signalling Pathway in Colorectal Cancer. Cells. 2023;12(3):447. [Google Scholar] [CrossRef] |
| 53. | Hu Y, Yan C, Mu L, Huang K, Li X, Tao D, et al. Fibroblast-Derived Exosomes Contribute to Chemoresistance through Priming Cancer Stem Cells in Colorectal Cancer. PLoS One. 2015;10(5):e0125625. [Google Scholar] [CrossRef] |
| 54. | Zhao H, Yan C, Hu Y, Mu L, Huang K, Li Q, et al. Sphere-forming assay vs. organoid culture: Determining long-term stemness and the chemoresistant capacity of primary colorectal cancer cells. Int J Oncol. 2019;54(3):893-904. [Google Scholar] [CrossRef] |
| 55. | Li X, Yan X, Wang Y, Kaur B, Han H, Yu J. The Notch signaling pathway: a potential target for cancer immunotherapy. J Hematol Oncol. 2023;16(1):45. [Google Scholar] [CrossRef] |
| 56. | Zhou B, Lin W, Long Y, Yang Y, Zhang H, Wu K, et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct Target Ther. 2022;7(1):95. [Google Scholar] [CrossRef] |
| 57. | Liao W, Zhang L, Chen X, Xiang J, Zheng Q, Chen N, et al. Targeting cancer stem cells and signalling pathways through phytochemicals: A promising approach against colorectal cancer. Phytomedicine. 2023;108:154524. [Google Scholar] [CrossRef] |
| 58. | Ma J, Gong Y, Sun X, Liu C, Li X, Sun Y, et al. Tumor suppressor FRMD3 controls mammary epithelial cell fate determination via notch signaling pathway. Sci Adv. 2024;10(27):eadk8958. [Google Scholar] [CrossRef] |
| 59. | Wang M, Tang L, Chen S, Wang L, Wu J, Zhong C, et al. ZNF217-activated Notch signaling mediates sulforaphane-suppressed stem cell properties in colorectal cancer. J Nutr Biochem. 2024;125:109551. [Google Scholar] [CrossRef] |
| 60. | Brisset M, Mehlen P, Meurette O, Hollande F. Notch receptor/ligand diversity: contribution to colorectal cancer stem cell heterogeneity. Front Cell Dev Biol. 2023;11:1231416. [Google Scholar] [CrossRef] |
| 61. | Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137(6):1124-1135. [Google Scholar] [CrossRef] |
| 62. | Ziskin JL, Dunlap D, Yaylaoglu M, Fodor IK, Forrest WF, Patel R, et al. In situ validation of an intestinal stem cell signature in colorectal cancer. Gut. 2013;62(7):1012-1023. [Google Scholar] [CrossRef] |
| 63. | Merchant AA, Matsui W. Targeting Hedgehog--a cancer stem cell pathway. Clin Cancer Res. 2010;16(12):3130-3140. [Google Scholar] [CrossRef] |
| 64. | Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8(2):97-106. [Google Scholar] [CrossRef] |
| 65. | Jing J, Wu Z, Wang J, Luo G, Lin H, Fan Y, et al. Hedgehog signaling in tissue homeostasis, cancers, and targeted therapies. Signal Transduct Target Ther. 2023;8(1):315. [Google Scholar] [CrossRef] |
| 66. | Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5(1):8. [Google Scholar] [CrossRef] |
| 67. | Kim BR, Na YJ, Kim JL, Jeong YA, Park SH, Jo MJ, et al. RUNX3 suppresses metastasis and stemness by inhibiting Hedgehog signaling in colorectal cancer. Cell Death Differ. 2020;27(2):676-694. [Google Scholar] [CrossRef] |
| 68. | Coni S, Infante P, Gulino A. Control of stem cells and cancer stem cells by Hedgehog signaling: pharmacologic clues from pathway dissection. Biochem Pharmacol. 2013;85(5):623-628. [Google Scholar] [CrossRef] |
| 69. | Brown Y, Hua S, Tanwar PS. Extracellular matrix-mediated regulation of cancer stem cells and chemoresistance. Int J Biochem Cell Biol. 2019;109:90-104. [Google Scholar] [CrossRef] |
| 70. | Zhu R, Gires O, Zhu L, Liu J, Li J, Yang H, et al. TSPAN8 promotes cancer cell stemness via activation of sonic Hedgehog signaling. Nat Commun. 2019;10(1):2863. [Google Scholar] [CrossRef] |
| 71. | Gu Y, Wang Y, He L, Zhang J, Zhu X, Liu N, et al. Circular RNA circIPO11 drives self-renewal of liver cancer initiating cells via Hedgehog signaling. Mol Cancer. 2021;20(1):132. [Google Scholar] [CrossRef] |
| 72. | Do AD, Wu KS, Chu SS, Giang LH, Lin YL, Chang CC, et al. LOXL1-AS1 contributes to metastasis in sonic-hedgehog medulloblastoma by promoting cancer stem-like phenotypes. J Exp Clin Cancer Res. 2024;43(1):130. [Google Scholar] [CrossRef] |
| 73. | Chen H, Nio K, Yamashita T, Okada H, Li R, Suda T, et al. BMP9-ID1 signaling promotes EpCAM-positive cancer stem cell properties in hepatocellular carcinoma. Mol Oncol. 2021;15(8):2203-2218. [Google Scholar] [CrossRef] |
| 74. | Kirilly D, Spana EP, Perrimon N, Padgett RW, Xie T. BMP signaling is required for controlling somatic stem cell self-renewal in the Drosophila ovary. Dev Cell. 2005;9(5):651-662. [Google Scholar] [CrossRef] |
| 75. | Ram Makena M, Gatla H, Verlekar D, Sukhavasi S, M KP, K CP. Wnt/beta-Catenin Signaling: The Culprit in Pancreatic Carcinogenesis and Therapeutic Resistance. Int J Mol Sci. 2019;20(17):4242. [Google Scholar] [CrossRef] |
| 76. | Degirmenci B, Valenta T, Dimitrieva S, Hausmann G, Basler K. GLI1-expressing mesenchymal cells form the essential Wnt-secreting niche for colon stem cells. Nature. 2018;558(7710):449-453. [Google Scholar] [CrossRef] |
| 77. | Toh TB, Lim JJ, Chow EK. Epigenetics in cancer stem cells. Mol Cancer. 2017;16(1):29. [Google Scholar] [CrossRef] |
| 78. | Yan G, Dai M, Zhang C, Poulet S, Moamer A, Wang N, et al. TGFbeta/cyclin D1/Smad-mediated inhibition of BMP4 promotes breast cancer stem cell self-renewal activity. Oncogenesis. 2021;10(3):21. [Google Scholar] [CrossRef] |
| 79. | Hernandez-Vargas H, Sincic N, Ouzounova M, Herceg Z. Epigenetic signatures in stem cells and cancer stem cells. Epigenomics. 2009;1(2):261-280. [Google Scholar] [CrossRef] |
| 80. | Freire NH, Jaeger MDC, de Farias CB, Nor C, Souza BK, Gregianin L, et al. Targeting the epigenome of cancer stem cells in pediatric nervous system tumors. Mol Cell Biochem. 2023;478(10):2241-2255. [Google Scholar] [CrossRef] |
| 81. | LaFave LM, Kartha VK, Ma S, Meli K, Del Priore I, Lareau C, et al. Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell. 2020;38(2):212-228.e13. [Google Scholar] [CrossRef] |
| 82. | Marquardt JU, Factor VM, Thorgeirsson SS. Epigenetic regulation of cancer stem cells in liver cancer: current concepts and clinical implications. J Hepatol. 2010;53(3):568-577. [Google Scholar] [CrossRef] |
| 83. | Wang Y, Hu P, Wang F, Xi S, Wu S, Sun L, et al. UHRF1 inhibition epigenetically reprograms cancer stem cells to suppress the tumorigenic phenotype of hepatocellular carcinoma. Cell Death Dis. 2023;14(6):381. [Google Scholar] [CrossRef] |
| 84. | Zeng Z, Fu M, Hu Y, Wei Y, Wei X, Luo M. Regulation and signaling pathways in cancer stem cells: implications for targeted therapy for cancer. Mol Cancer. 2023;22(1):172. [Google Scholar] [CrossRef] |
| 85. | Wang Q, Liang N, Yang T, Li Y, Li J, Huang Q, et al. DNMT1-mediated methylation of BEX1 regulates stemness and tumorigenicity in liver cancer. J Hepatol. 2021;75(5):1142-1153. [Google Scholar] [CrossRef] |
| 86. | Liu S, Cheng K, Zhang H, Kong R, Wang S, Mao C, et al. Methylation Status of the Nanog Promoter Determines the Switch between Cancer Cells and Cancer Stem Cells. Adv Sci (Weinh). 2020;7(5):1903035. [Google Scholar] [CrossRef] |
| 87. | Zagorac S, Alcala S, Fernandez Bayon G, Bou Kheir T, Schoenhals M, Gonzalez-Neira A, et al. DNMT1 Inhibition Reprograms Pancreatic Cancer Stem Cells via Upregulation of the miR-17-92 Cluster. Cancer Res. 2016;76(15):4546-4558. [Google Scholar] [CrossRef] |
| 88. | French R, Pauklin S. Epigenetic regulation of cancer stem cell formation and maintenance. Int J Cancer. 2021;148(12):2884-2897. [Google Scholar] [CrossRef] |
| 89. | Castilho RM, Squarize CH, Almeida LO. Epigenetic Modifications and Head and Neck Cancer: Implications for Tumor Progression and Resistance to Therapy. Int J Mol Sci. 2017;18(7):1506. [Google Scholar] [CrossRef] |
| 90. | Yasuda H, Soejima K, Watanabe H, Kawada I, Nakachi I, Yoda S, et al. Distinct epigenetic regulation of tumor suppressor genes in putative cancer stem cells of solid tumors. Int J Oncol. 2010;37(6):1537-1546. [Google Scholar] [CrossRef] |
| 91. | Schniewind I, Hadiwikarta WW, Grajek J, Poleszczuk J, Richter S, Peitzsch M, et al. Cellular plasticity upon proton irradiation determines tumor cell radiosensitivity. Cell Rep. 2022;38(8):110422. [Google Scholar] [CrossRef] |
| 92. | Perusina Lanfranca M, Thompson JK, Bednar F, Halbrook C, Lyssiotis C, Levi B, et al. Metabolism and epigenetics of pancreatic cancer stem cells. Semin Cancer Biol. 2019;57:19-26. [Google Scholar] [CrossRef] |
| 93. | Galassi C, Esteller M, Vitale I, Galluzzi L. Epigenetic control of immunoevasion in cancer stem cells. Trends Cancer. 2024 Nov;10(11):1052-1071. [Google Scholar] [CrossRef] |
| 94. | Wang X, Li N, Zheng M, Yu Y, Zhang S. Acetylation and deacetylation of histone in adipocyte differentiation and the potential significance in cancer. Transl Oncol. 2024;39:101815. [Google Scholar] [CrossRef] |
| 95. | Xu X, Ding Y, Jin J, Xu C, Hu W, Wu S, et al. Post-translational modification of CDK1-STAT3 signaling by fisetin suppresses pancreatic cancer stem cell properties. Cell Biosci. 2023;13(1):176. [Google Scholar] [CrossRef] |
| 96. | Bi L, Ren Y, Feng M, Meng P, Wang Q, Chen W, et al. HDAC11 Regulates Glycolysis through the LKB1/AMPK Signaling Pathway to Maintain Hepatocellular Carcinoma Stemness. Cancer Res. 2021;81(8):2015-2028. [Google Scholar] [CrossRef] |
| 97. | Caslini C, Hong S, Ban YJ, Chen XS, Ince TA. HDAC7 regulates histone 3 lysine 27 acetylation and transcriptional activity at super-enhancer-associated genes in breast cancer stem cells. Oncogene. 2019;38(39):6599-6614. [Google Scholar] [CrossRef] |
| 98. | Zhou X, Ao X, Jia Z, Li Y, Kuang S, Du C, et al. Non-coding RNA in cancer drug resistance: Underlying mechanisms and clinical applications. Front Oncol. 2022;12:951864. [Google Scholar] [CrossRef] |
| 99. | Liu S, Sun Y, Hou Y, Yang L, Wan X, Qin Y, et al. A novel lncRNA ROPM-mediated lipid metabolism governs breast cancer stem cell properties. J Hematol Oncol. 2021;14(1):178. [Google Scholar] [CrossRef] |
| 100. | He Y, Jiang X, Duan L, Xiong Q, Yuan Y, Liu P, et al. LncRNA PKMYT1AR promotes cancer stem cell maintenance in non-small cell lung cancer via activating Wnt signaling pathway. Mol Cancer. 2021;20(1):156. [Google Scholar] [CrossRef] |
| 101. | Yan H, Bu P. Non-coding RNAs in cancer stem cells. Cancer Lett. 2018;421:121-126. [Google Scholar] [CrossRef] |
| 102. | Ren J, Ding L, Zhang D, Shi G, Xu Q, Shen S, et al. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics. 2018;8(14):3932-3948. [Google Scholar] [CrossRef] |
| 103. | Feng Y, Cai L, Pook M, Liu F, Chang CH, Mouti MA, et al. BRD9-SMAD2/3 Orchestrates Stemness and Tumorigenesis in Pancreatic Ductal Adenocarcinoma. Gastroenterology. 2024;166(1):139-154. [Google Scholar] [CrossRef] |
| 104. | Deshmukh A, Binju M, Arfuso F, Newsholme P, Dharmarajan A. Role of epigenetic modulation in cancer stem cell fate. Int J Biochem Cell Biol. 2017;90:9-16. [Google Scholar] [CrossRef] |
| 105. | Feng Y, Liu X, Pauklin S. 3D chromatin architecture and epigenetic regulation in cancer stem cells. Protein Cell. 2021;12(6):440-454. [Google Scholar] [CrossRef] |
| 106. | Rajabi A, Kayedi M, Rahimi S, Dashti F, Mirazimi SMA, Homayoonfal M, et al. Non-coding RNAs and glioma: Focus on cancer stem cells. Mol Ther Oncolytics. 2022;27:100-123. [Google Scholar] [CrossRef] |
| 107. | Ghasemi S, Xu S, Nabavi SM, Amirkhani MA, Sureda A, Tejada S, et al. Epigenetic targeting of cancer stem cells by polyphenols (cancer stem cells targeting). Phytother Res. 2021;35(7):3649-3664. [Google Scholar] [CrossRef] |
| 108. | Portney BA, Arad M, Gupta A, Brown RA, Khatri R, Lin PN, et al. ZSCAN4 facilitates chromatin remodeling and promotes the cancer stem cell phenotype. Oncogene. 2020;39(26):4970-4982. [Google Scholar] [CrossRef] |
| 109. | Yamashita N, Kufe D. Addiction of Cancer Stem Cells to MUC1-C in Triple-Negative Breast Cancer Progression. Int J Mol Sci. 2022;23(15):8219. [Google Scholar] [CrossRef] |
| 110. | Zhou L, Wen R, Bai C, Li Z, Zheng K, Yu Y, et al. Spatial transcriptomic revealed intratumor heterogeneity and cancer stem cell enrichment in colorectal cancer metastasis. Cancer Lett. 2024;602:217181. [Google Scholar] [CrossRef] |
| 111. | Ma Y, He L, Zhao X, Li W, Lv X, Zhang X, et al. Protease activated receptor 2 signaling promotes self-renewal and metastasis in colorectal cancer through beta-catenin and periostin. Cancer Lett. 2021;521:130-141. [Google Scholar] [CrossRef] |
| 112. | Wang D, Fu L, Sun H, Guo L, DuBois RN. Prostaglandin E2 Promotes Colorectal Cancer Stem Cell Expansion and Metastasis in Mice. Gastroenterology. 2015;149(7):1884-1895.e4. [Google Scholar] [CrossRef] |
| 113. | Gavert N, Vivanti A, Hazin J, Brabletz T, Ben-Ze'ev A. L1-mediated colon cancer cell metastasis does not require changes in EMT and cancer stem cell markers. Mol Cancer Res. 2011;9(1):14-24. [Google Scholar] [CrossRef] |
| 114. | de Sousa e Melo F, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J, et al. A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature. 2017;543(7647):676-680. [Google Scholar] [CrossRef] |
| 115. | Gerstberger S, Jiang Q, Ganesh K. Metastasis. Cell. 2023;186(8):1564-1579. [Google Scholar] [CrossRef] |
| 116. | Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. 2014;14(3):342-356. [Google Scholar] [CrossRef] |
| 117. | Li R, Liu X, Huang X, Zhang D, Chen Z, Zhang J, et al. Single-cell transcriptomic analysis deciphers heterogenous cancer stem-like cells in colorectal cancer and their organ-specific metastasis. Gut. 2024;73(3):470-484. [Google Scholar] [CrossRef] |
| 118. | Li Y, Wang Z, Ajani JA, Song S. Drug resistance and Cancer stem cells. Cell Commun Signal. 2021;19(1):19. [Google Scholar] [CrossRef] |
| 119. | Alketbi L, Al-Ali A, Talaat IM, Hamid Q, Bajbouj K. The Role of ATP-Binding Cassette Subfamily A in Colorectal Cancer Progression and Resistance. Int J Mol Sci. 2023;24(2):1344. [Google Scholar] [CrossRef] |
| 120. | Chen W, Zhang Q, Dai X, Chen X, Zhang C, Bai R, et al. PGC-1alpha promotes colorectal carcinoma metastasis through regulating ABCA1 transcription. Oncogene. 2023;42(32):2456-2470. [Google Scholar] [CrossRef] |
| 121. | Zhu Y, Huang S, Chen S, Chen J, Wang Z, Wang Y, et al. SOX2 promotes chemoresistance, cancer stem cells properties, and epithelial-mesenchymal transition by beta-catenin and Beclin1/autophagy signaling in colorectal cancer. Cell Death Dis. 2021;12(5):449. [Google Scholar] [CrossRef] |
| 122. | Mangiapane LR, Nicotra A, Turdo A, Gaggianesi M, Bianca P, Di Franco S, et al. PI3K-driven HER2 expression is a potential therapeutic target in colorectal cancer stem cells. Gut. 2022;71(1):119-128. [Google Scholar] [CrossRef] |
| 123. | Yuan Z, Liang X, Zhan Y, Wang Z, Xu J, Qiu Y, et al. Targeting CD133 reverses drug-resistance via the AKT/NF-kappaB/MDR1 pathway in colorectal cancer. Br J Cancer. 2020;122(9):1342-1353. [Google Scholar] [CrossRef] |
| 124. | Li J, Wu C, Hu H, Qin G, Wu X, Bai F, et al. Remodeling of the immune and stromal cell compartment by PD-1 blockade in mismatch repair-deficient colorectal cancer. Cancer Cell. 2023;41(6):1152-1169.e7. [Google Scholar] [CrossRef] |
| 125. | Sole L, Lobo-Jarne T, Alvarez-Villanueva D, Alonso-Maranon J, Guillen Y, Guix M, et al. p53 wild-type colorectal cancer cells that express a fetal gene signature are associated with metastasis and poor prognosis. Nat Commun. 2022;13(1):2866. [Google Scholar] [CrossRef] |
| 126. | Dong S, Liang S, Cheng Z, Zhang X, Luo L, Li L, et al. ROS/PI3K/Akt and Wnt/beta-catenin signalings activate HIF-1alpha-induced metabolic reprogramming to impart 5-fluorouracil resistance in colorectal cancer. J Exp Clin Cancer Res. 2022;41(1):15. [Google Scholar] [CrossRef] |
| 127. | Woolston A, Khan K, Spain G, Barber LJ, Griffiths B, Gonzalez-Exposito R, et al. Genomic and Transcriptomic Determinants of Therapy Resistance and Immune Landscape Evolution during Anti-EGFR Treatment in Colorectal Cancer. Cancer Cell. 2019;36(1):35-50 e39. [Google Scholar] [CrossRef] |
| 128. | Li Y, Mu L, Li Y, Mi Y, Hu Y, Li X, et al. Golgi dispersal in cancer stem cells promotes chemoresistance of colorectal cancer via the Golgi stress response. Cell Death Dis. 2024;15(6):417. [Google Scholar] [CrossRef] |

![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy




