
Cancer Heterogeneity and Plasticity ISSN 2818-7792
Cancer Heterogeneity and Plasticity 2025;2(2):0006 | https://doi.org/10.47248/chp2502020006
Review Open Access
Lipid Metabolism and Immune Response in Hepatocellular Carcinoma: Interplay Driving Tumor Progression
Tiffany Ching-Yun Yu
1,2,†

 ,
Yu-Man Tsui
1,2,†
,
Vanilla Xin Zhang
1,2,†
,
Huanhuan Ma
1,2
,
Irene Oi-Lin Ng
1,2
,
Yu-Man Tsui
1,2,†
,
Vanilla Xin Zhang
1,2,†
,
Huanhuan Ma
1,2
,
Irene Oi-Lin Ng
1,2
Correspondence: Yu-Man Tsui; Irene Oi-Lin Ng
Academic Editor(s): Dean G. Tang
Received: Dec 6, 2024 | Accepted: Mar 25, 2025 | Published: Apr 4, 2025
© 2025 by the author(s). This is an Open Access article distributed under the terms of the Creative Commons License Attribution 4.0 International (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: Yu TC-Y, Tsui Y-M, Zhang VX, Ma H, Ng IO-L. Lipid Metabolism and Immune Response in Hepatocellular Carcinoma: Interplay Driving Tumor Progression. Cancer Heterog Plast. 2025;2(2):0006. https://doi.org/10.47248/chp2502020006
With the rising incidence of metabolic dysfunction-associated steatotic liver disease (MASLD), it has become a significant risk factor for hepatocellular carcinoma (HCC). This review focuses on the roles of lipid metabolism aberrations and reprogramming in HCC development. We begin with a brief overview of the relevant lipids to HCC, including fatty acyls, glycerolipids, glycerophospholipids and sterol lipids, and discuss particularly how the associated lipid metabolism and its reprogramming promotes chemoresistance in HCC. We then explore the heterogeneity in lipid distribution and metabolism across different stages of HCC development. This includes intra-tissue spatial heterogeneity across histological structure and zonated regions in the liver, and interpatient tumor heterogeneity at various degrees of resolutions, from single cell to bulk tissue levels. Next, we describe the plasticity in lipid metabolism in MASLD and HCC. With the advent of immunotherapy for HCC, we also examine the relationship between lipid metabolism and anti-tumor immunity in HCC. Finally, we address the challenges and future perspectives of targeting lipid metabolism and tumor immunity as a dual approach to improve HCC treatment.
KeywordsLipid metabolism, tumor immunity, hepatocellular carcinoma, heterogeneity, chemoresistance
Hepatocellular carcinoma (HCC) is a prevalent malignancy worldwide, with high incidence rates in Asia due to hepatitis virus infection [1]. Recently, the incidence of metabolic dysfunction-associated steatotic liver disease (MASLD) is rising globally and it has become a significant risk factor for HCC [1,2]. Lipid metabolism has garnered increasing attention because lipids are crucial components of cellular membranes and serve as energy storage, which is essential for rapidly proliferating cancer cells. Lipid conjugation to proteins enables these proteins to anchor to the cell membrane lipid bilayer [3], influencing membrane fluidity and modulating signal transduction, thereby regulating various cellular properties and functions [4,5].
As the liver plays a critical role in lipid metabolism, including lipid synthesis, storage, and breakdown to maintain lipid homeostasis [6], understanding the relationship between lipid metabolism and HCC development has become a major research focus. Studies have shown that HCC cells reprogram fatty acid metabolism through epigenetic and transcriptional regulation [7,8], allowing them to use lipids as an energy source under nutrient-deprived conditions. This metabolic reprogramming demonstrates the plasticity of cancer cells - the ability to acquire phenotypes of other cell types in a reversible manner [9], including metabolic plasticity, trans-differentiation, dedifferentiation, and epithelial-mesenchymal transition (EMT) [9].
Ongoing research aims to identify lipid metabolism-related gene markers for prognosis and therapeutic targets in HCC [10]. With advancements in immunotherapy in HCC, exploring how targeting lipid metabolism can be integrated with immunotherapy presents an intriguing area for developing future therapeutic strategies. A comprehensive understanding of lipid metabolism reprogramming in HCC, particularly in relation to chemoresistance, heterogeneity, plasticity and anti-tumor immunity, is essential.
In this review, we will first describe general lipid metabolism in HCC, followed by discussions on reprogramming, chemoresistance, heterogeneity and plasticity issues related to lipid metabolism in HCC. We will also examine how lipids or lipid metabolism impact cellular plasticity in HCC. After a brief introduction to tumor immunity, we will elaborate on the relationship between lipid metabolism and anti-tumor immunity in HCC. Finally, we will address the challenges and future perspectives of targeting lipid metabolism and tumor immunity as a dual approach to improve HCC treatment.
Lipids are a diverse group of hydrophobic metabolites essential for various cellular functions. According to the LIPID metabolites and pathways strategy consortium [11], lipids can be categorized into eight groups. In this review, we focus on the lipid categories most extensively studied and reported in the context of HCC: fatty acyls, glycerolipids, glycerophospholipids and sterol lipids.
Fatty acids (FAs) in the liver are derived from either de novo lipogenesis (DNL) or uptake of plasma non-esterified fatty acids (NEFAs) [12]. In DNL, acetyl-CoA carboxylase (ACC) synthesizes acyl-CoA, which is then extended by fatty acid synthase (FASN) [12] (Figure 1). This process is followed by desaturation and elongation by stearoyl-CoA desaturases (SCDs) and very-long-chain fatty acid elongases (ELOVLs) [13]. Plasma NEFAs are transported into hepatocytes via CD36, SLC27, other membrane FA-binding proteins (FABPs), or simple diffusion [12]. Once inside the mitochondria, FAs are transported through a carnitine-dependent system involving carnitine palmitoyltransferases (CPT1 and CPT2) and SLC25A20. There, they are activated and oxidized for ATP synthesis, generating acetyl-CoA [12,14]. Additionally, FAs serve as building blocks for lipid mediators like eicosanoids [11] or binds to transcription factors to drive regulatory and inflammatory signaling [15].

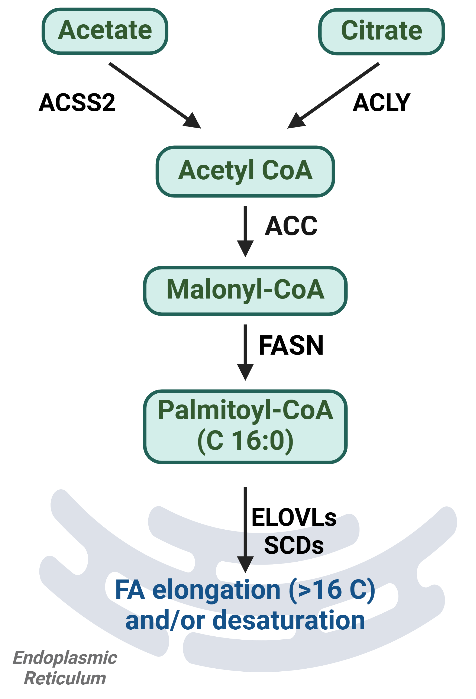
Figure 1. Summary diagram for de novo lipogenesis pathway. Fatty acids are synthesized from acetate and citrate through a series of intermediates, with various metabolic enzymes catalyzing each step in the lipogenesis pathway. ACSS2, acyl-CoA synthetase short chain family member 2; ACLY, ATP citrate lyase; ACC, acetyl-CoA carboxylase; FASN, fatty acid synthase; ELOVLs, very-long-chain fatty acid elongases; SCDs, stearoyl-CoA desaturases. Figure was created by BioRender.com.
Glycerolipids, which include tri-, di-, and mono-acylglycerols (TAGs, DAGs and MAGs), contain FAs esterified to a glycerol backbone [11]. Their synthesis (Figure 2) involves key enzymes such as glycerol-3-phosphate acyltransferases (GPATs), acylglycerol-3-phosphate acyltransferases (AGPATs), DAG-acyltransferase (DGATs), and lipins [16,17]. MAGs regenerate TAG through the monoacylglycerol acyltransferase (MGAT) pathway [18]. TAGs are then packaged with cholesteryl esters into lipid droplets (LDs) for intracellular storage and mobilization [19]. At sites of demand, TAGs can be hydrolyzed by lipases (e.g. adipocyte triglyceride lipase (ATGL), MAG lipase (MAGL)) or consumed via lipophagy [20]. LD surface proteins (e.g. perilipins [PLINs]) and carboxylesterase 1 and 2 (CES1/2) further regulate LD mobilization [20] and TAG hydrolysis [21].
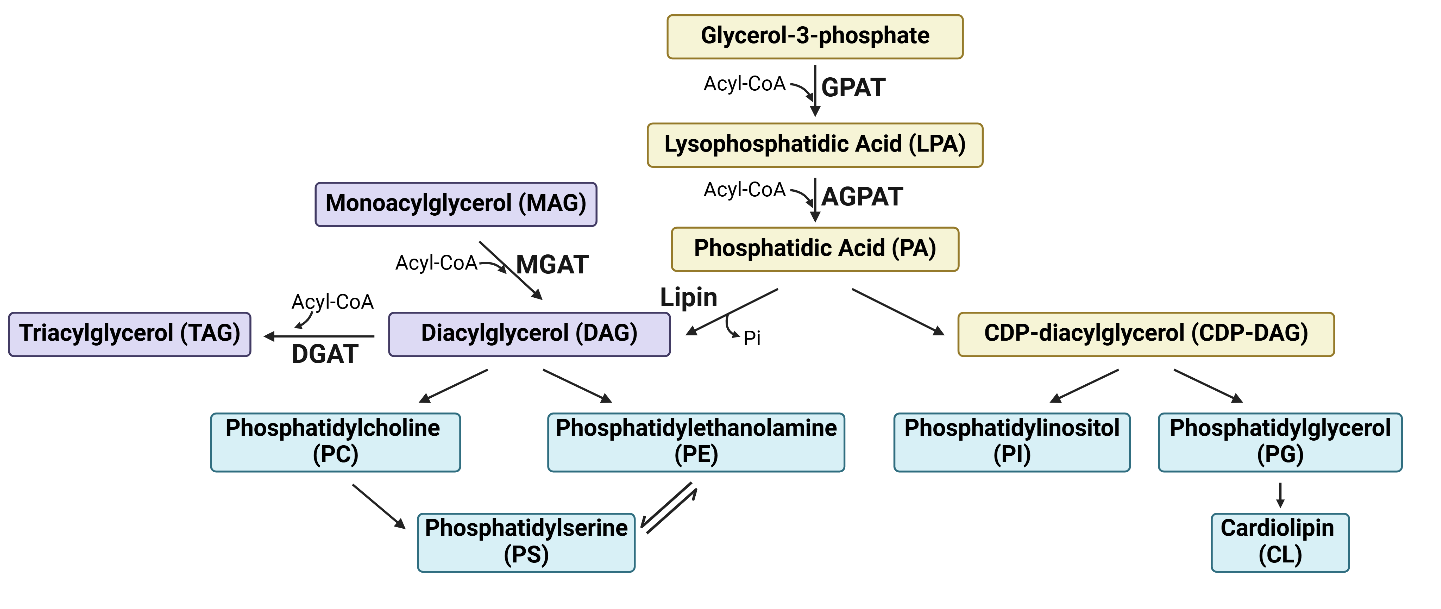
Figure 2. Summary diagram for glycerolipids and glycerophospholipids synthesis. Glycerol-3-phosphate (G3P) serves as the precursor of glycerolipids and glycerophospholipids. G3P is first converted to phosphatidic acid (PA) through the sequential addition of acyl-CoA molecules by the enzymes GPATs and AGPATs. PA then serves as a key intermediate for the synthesis of various glycerolipids (shaded in purple) and glycerophospholipids (shaded in blue). GPAT, glycerol-3-phosphate acyltransferase; AGPAT, acylglycerol-3-phosphate acyltransferase; MGAT, monoacylglycerol acyltransferase; DGAT, diacylglycerol-acyltransferase; CDP, Cytidine diphosphate. Figure was created by BioRender.com.
Hence, TAGs function as energy storage and homeostasis, providing a critical source of FAs during energy deprivation. They also sequester PUFAs and toxic saturated FAs to prevent oxidative stresses, ferroptosis and lipotoxicity [22–26]. DAGs, as the intermediates in glycerolipid metabolism, serve as precursors for glycerophospholipids (discussed below). Lastly, DAGs and PAs are also secondary messengers to affect multiple cellular functions [27–29].
Glycerophospholipids (GPs), which are synthesized from DAG or CDP-DAG (Figure 2), are essential components of cellular membranes [11,30]. The acyl chains in GPs can be further modified by exchanging them with FAs of different lengths and degrees of saturation through Lands’ cycle. This process results in changes to membrane fluidity and stiffness, thereby influencing various biological processes [31]. Additionally, phosphatidylinositol (PtdIns), plays a role in intracellular calcium homeostasis and signaling by being hydrolyzed by phospholipase to generate inositol-1,4,5-trisphosphate [29].
Sterols, including cholesterol and its derivatives, are obtained from both exogenous (dietary) source and endogenous biogenesis [11]. Hepatocytes absorb dietary cholesterol from chylomicron remnants [32], while de novo cholesterol synthesis occurs in the liver from acetyl-CoA via the mevalonate pathway, with acetyl-CoA as the precursor (Figure 3). Key rate-limiting enzymes in this pathway include HMG-CoA reductase (HMGCR) and squalene epoxidase (SQLE) [33,34]. Cholesterol is esterified with FAs by sterol O-acyltransferases (SOATs) and stored in LDs along with TAGs [33]. Free cholesterol is transported out of cells by ATP binding cassette transporters (e.g. ABCA1, ABCG1) to form high- and very low-density lipoprotein (HDL and VLDL) [33]. Cholesterol also serves as a precursor for steroid hormones and bile acids [11]. Additionally, the incorporation of cholesterol into cellular membranes decreases membrane fluidity, which affects lipid raft formation and regulates certain membrane proteins, ligands and receptors, thereby modulating cellular communications [35,36].
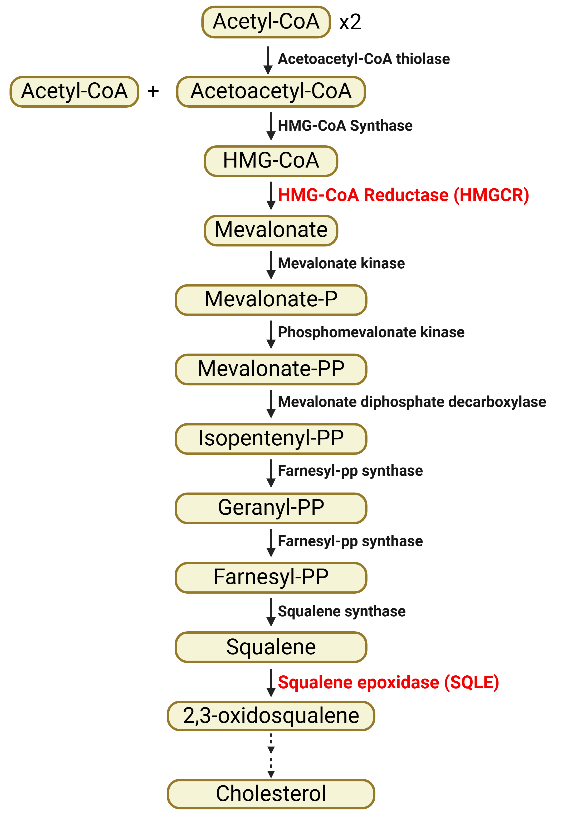
Figure 3. Summary diagram for de novo cholesterol synthesis via mevalonate pathway. Squalene, produced from two acetyl-CoA molecules through a series of enzymatic conversions, is used in cholesterol synthesis. P, phosphate; PP, pyrophosphate. Red-highlighted are key rate-limiting enzymes. Figure was created by BioRender.com.
Extensive studies on lipid metabolism in HCC have been reviewed in multiple articles [3,34,37–40]. Here, we briefly summarize the reprogramming of FAs, cholesterol, TAGs and LD and GP metabolism (Figure 4).
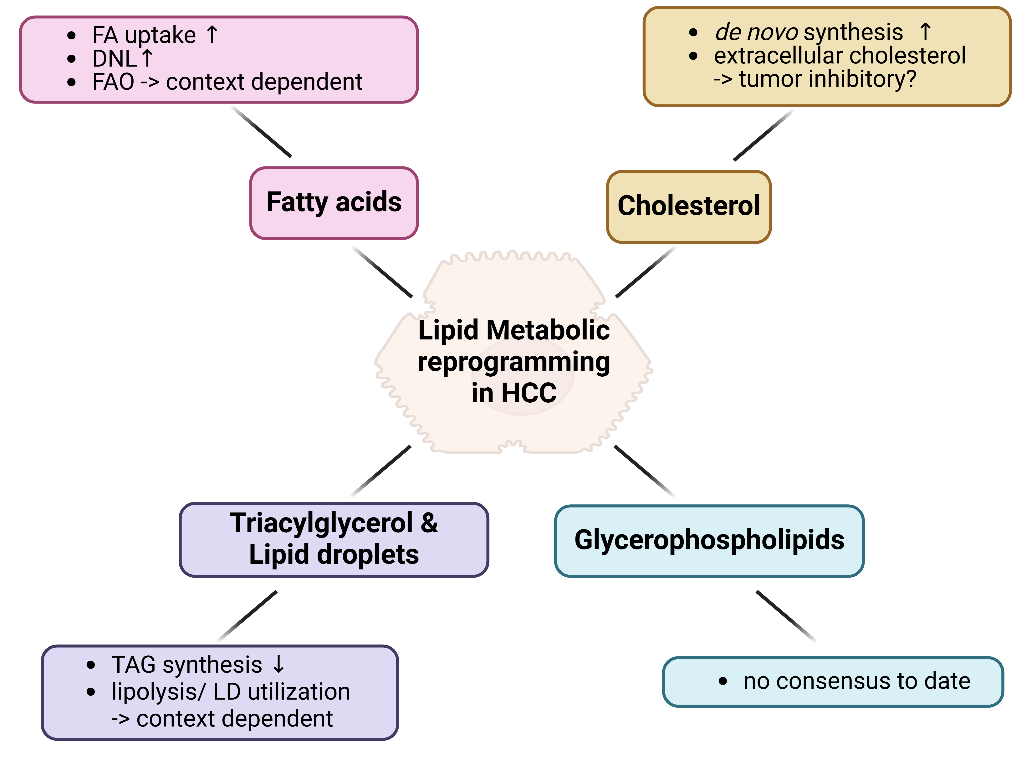
Figure 4. Summary for lipid metabolic reprogramming in HCC. Lipid metabolic reprogramming in HCC is categorized into four main aspects based on the types of lipid metabolites: fatty acids, cholesterol, triacylglycerols and lipid droplets, and glycerophospholipids. Each category is elaborated in the respective sections. Figure was created by BioRender.com.
Multiomics analysis has indicated FA accumulation in HCC [39,41], which can be attributed to changes in FA uptake, DNL and fatty acid oxidation (FAO). For instance, CD36, which facilitates FA uptake, is upregulated in HCC and supports tumor initiation and EMT and metastasis [39,41]. Additionally, SLC27A4 selectively absorbs monounsaturated FA (MUFAs) to resist sorafenib-induced ferroptosis in HCC [42].
Key enzymes involved in DNL, such as ATP citrate lyase (ACLY), ACC, and FASN are upregulated in HCC and have been shown to support tumor stemness, growth and metastasis in vitro and in vivo [37,41]. Stearoyl-CoA desaturase 1 (SCD1), which facilitates FA desaturation, is also activated in HCC and is enhanced by matrix stiffness during liver cirrhosis to promote metastasis by reducing membrane fluidity [39,43]. SREBPs, the master regulators of DNL, are influenced by c-Myc and PI3K/Akt/mTOR signaling, which are frequently activated in HCC. Crosstalk with glutamine metabolism also supports lipogenesis via SREBPs, promoting tumor formation and progression [44,45].
The role of FAO in HCC, however, remains controversial. For example, CPT1/2 and SLC25A20 are downregulated in HCC, inhibiting long-chain FA oxidation [37,41]. Hypoxic condition can also inhibit FAO via HIF-1α suppression on medium- and long-chain acyl-CoA dehydrogenase (MCAD and LCAD) expression [37], suggesting an inhibitory role of FAO in HCC. Conversely, some studies suggest FAO promotes tumor growth; nutrient deprivation-induced mitochondrial translocation of CPT1A and β-catenin-enhanced expression of PPARα, CPT2, MCAD, and LCAD promote FAO in HCC [37]. Moreover, acyl-CoA synthetase long chain family member 4, which oxidizes long-chain FA released from membrane GPs and supports ferroptosis, paradoxically promotes HCC proliferation [46]. Thus, the role of FAO in HCC is likely context dependent.
Cholesterol de novo synthesis is often enhanced in HCC and is suggested to promote tumor growth. HMGCR, a key enzyme in this pathway, is upregulated in HCC, and statins, which inhibit HMGCR, have been shown to reduce liver cancer risk [41]. Additionally, knockout of miR-148a, which normally inhibits HMGCR expression, increases cholesterol synthesis and enhances diethylnitrosamine (DEN)-induced tumorigenesis in mice, further supporting a positive role of cholesterol in tumor promotion [47]. Upregulation of SQLE and sterol O-acyltransferase 1 (SOAT1) is associated with worse prognosis and functionally supports tumor growth, EMT, and metastasis in HCC [41,48]. Interestingly, exogenous cholesterol uptake may have opposite effects. Low levels of plasma cholesterol, particularly HDL, correlate with worse survival and more aggressive clinical features [49–51]. Cholesterol treatment has been shown to reduce HCC proliferation and metastasis by inhibiting sterol regulatory element-binding protein cleavage-activating protein, thereby reducing de novo lipogenesis (DNL) [52]. This underscores the coordination between DNL and cholesterol synthesis, further exemplified by hypoxia-induced activation of the SREBP1-ACLY axis to promote cholesterol accumulation in HCC [40].
TAG synthesis has been suggested to have tumor-suppressive effects. DGAT2, which catalyzes the final step in TAG synthesis, is downregulated in clinical HCC samples and has been shown to block the G1-S transition, thereby reducing HCC proliferation [53]. Conversely, DGAT1 deficiency has been found to enhance stem-cell properties, promote proliferation, and induce EMT in HCC cell lines [53].
The role of TAG utilization from LDs and its regulation by LD surface proteins remain unclear. In non-alcoholic fatty liver disease (NAFLD) (now named MALSD)-HCC, PLIN3 promotes cancer metastasis, while PLIN5 binds to ATGL to inhibit lipolysis, promoting steatosis and driving MASLD-HCC formation [54–56]. Supporting the hypothesis that lipid utilization is tumor-promoting during nutrient deficiency, glucose starvation leads to phosphorylation of the glycolytic enzyme phosphofructokinase of the liver type, repurposing it as a protein kinase to facilitate LD mitochondrial tethering and downstream utilization through PLIN2-CPT1A interaction [57].
In line with this, ATGL has been shown to promote tumor proliferation in HCC by providing metabolic products such as DAGs and NEFAs [58,59], while also reducing intracellular triglycerides and promoting resistance to lipotoxicity [60]. Moreover, MAGL, another lipase involved in the hydrolysis of glycerolipids in LDs, exerts a tumor-promoting effect through downstream metabolites prostaglandin E2 and lysophosphatidic acid (LPA) [61]. However, opposing findings on ATGL suggest a context-dependent role of lipolysis in HCC. Overexpression of ATGL has been shown to induce a metabolic switch from glycolysis to oxidative metabolism in mitochondria, suppressing tumor proliferation in specific HCC and hepatoma cell lines [62].
As GPs are major components of cell membranes, their amount and composition can influence tumor plasma membrane properties, exerting either tumor-promoting or tumor-suppressive effects depending on the context. Lysophosphatidylcholine acyltransferases (LPCATs), which convert lysophosphatidylcholine (lyso-PtdCho) to phosphatidylcholine (PtdCho), are upregulated in HCC [63]. This upregulation leads to an enrichment of PtdCho with palmitoleic or oleic acid and a reduction of lyso-PtdCho with these fatty acids. The accumulation of PtdCho supports tumor proliferation, migration, and invasion [64]. However, another study using phospholipid isotope tracing found reduced PtdCho levels in β-catenin-driven HCC [65]. LPCAT3, another enzyme involved in GP remodeling, was found to suppress mitochondrial GP saturation and subsequent autophagy, thereby inhibiting spontaneous and diet-induced NASH/HCC tumor formation [66]. Therefore, the role of GP modification in HCC progression remains unclear.
As previously mentioned, membrane phospholipids can also serve as sources of fatty acids for the synthesis of lipid mediators. One notable example is prostaglandin E2 (PGE2), which mediates inflammation and has been extensively studied for its role in HCC, as reviewed by Chen et.al [67]. In brief, cyclooxygenase-2 (COX2) is upregulated in HCC, and elevated PGE2 levels in the bloodstream correlate with larger tumors and poorer survival outcomes for HCC patients [67]. Mechanistically, autocrine and paracrine signaling through prostaglandin E receptors (EPs) on HCC cells supports tumor cell survival, growth, migration, and invasion [67].
Recent studies on HCC chemoresistance have identified several genes that also regulate lipid metabolism, as summarized in Table 1. In oxaliplatin-resistant HCC cells [39], proteins involved in lipid and energy metabolism were upregulated, indicating active lipid metabolism in these chemo-resistant cells. This was further supported by the increased expression of lipogenic genes (FAS, ACC, ACLY) and decreased expression of lipolytic genes (MCAD, LCAD, CPT2), leading to lipid accumulation in oxaliplatin-resistant HCC cells.
Table 1. Summary of genes of interest that have functional roles in chemoresistance and lipid or carbon metabolism in HCC.
Chromatin immunoprecipitation (ChIP) sequencing data for the transcription factor NANOG revealed an enrichment of lipid metabolism pathway-related genes and the physical binding of NANOG to the ACLY promoter [68]. This drives the expression of various genes related to fatty acid synthesis, suggesting that NANOG may sensitize tumor growth towards the inhibitory effects of sorafenib in HCC cells.
Recent research has explored the role of CES1 in HCC chemoresistance [69]. CES1, which facilitates TAG hydrolysis, was inhibited using WWL229, leading to altered lipid profiles and impaired mitochondrial function in HCC cells. CES1 knockout or inhibition suppressed lipid hydrolysis, causing lipid droplet accumulation and further impairing mitochondrial function [69]. There was a significant positive correlation between CES1 and SCD proteins, and the knockdown of PPARα or γ abolished CES1's ability to restore SCD expression. As SCD converts saturated fatty acids into MUFAs and PUFAs, CES1 inhibition or knockout reduced SCD1 protein levels, altering the lipid profiles of HCC cells [69]. CES1 inhibition also sensitized HCC cells to cisplatin, increasing apoptotic cell percentages compared to single drug treatments, and demonstrated a synergistic effect in tumor growth suppression in vivo. This suggests that the CES1-SCD axis links lipid profile alterations to HCC cell chemosensitivity.
In another study, downregulation of CPT2, a rate-limiting enzyme for FAO, was associated with poor differentiation and venous invasion in HCC [70]. CPT2 knockdown in HepG2 cells enhanced proliferation, colony formation, migration, and chemoresistance to cisplatin, supporting a tumor-suppressive role for CPT2 in HCC. CPT2 knockdown also promoted lipogenesis and enhanced SCD expression, increasing levels of TAG, cholesterol, and phospholipids. This suggests that CPT2 downregulation may promote hepatocarcinogenesis through increased MUFA levels by SCD [71]. Further validation is needed through rescue experiments using SCD suppression in CPT2-knockdown cells. In line with this, another study found that SCD plays a positive role in HCC proliferation and chemoresistance, with SCD being upregulated in HCC and associated with poorer differentiation. Chemotherapy drugs like 5-FU and doxorubicin increased SCD expression over time, and SCD knockdown or inhibition sensitized HCC cells to these drugs in an SREBP1-dependent manner [72].
Liver-specific knockout of the mRNA-binding protein tristetraprolin (TTP) reduced tumor burden in DEN-treated mouse HCC models, suggesting an oncogenic role for TTP in HCC [73]. Lipidomic profiling showed that TTP knockout promoted MUFAs and abolished short-term DEN-induced PUFAs. Overexpression of TTP sensitized HCC cells to doxorubicin and sorafenib, highlighting the role of lipid profile changes in chemoresistance modulated by TTP. These studies collectively suggest that reprogramming lipid metabolism plays a significant role in the chemoresistance of HCC cells.
NAFLD or MASLD is closely linked to HCC, with a clear causal connection to HCC of metabolic disorder etiology [74]. The progression to these types of HCC typically involves a gradual transition from metabolic disorders and fatty liver to severe manifestations such as cirrhosis and ultimately HCC. Various research groups have studied the heterogeneity of lipid metabolism at different stages of HCC development. Here, we discuss this progression from normal liver, through NAFLD and cirrhosis, to HCC.
In normal liver, the spatial distribution of lipids has been examined using desorption electrospray ionization-mass spectrometry imaging (DESI-MSI) at a resolution of 50μm x 50μm on mouse liver tissues [75]. This study identified statistically significant lipid distributions across liver zonation. Polyunsaturated fatty acids (PUFAs) such as FA (18:2), FA (20:2), FA (20:3), arachidonic acid [FA (20:4)], FA (22:4), and docosahexaenoic acid (DHA) [FA (22:6)], as well as certain phospholipids containing these acids, were enriched in periportal hepatocytes. Specific phosphatidylcholines (PtdCho) and phosphatidylinositols (PtdIns) displayed distinct distributions across periportal, midzone, or pericentral regions. While this study highlights intrahepatic lipidomic heterogeneity, its resolution only demonstrates spatial heterogeneity across histologically labeled structures and various zones of interest.
In contrast, another study used a DESI workflow on consecutive tissue sections from multiple mouse and human liver tissues at single-cell resolution to explore metabolic heterogeneity across liver zonation and functional units [76]. This study quantified the intensity of different metabolites and lipids down to specific cell types labeled with cell type-specific markers. It found that different cell types were enriched with distinct metabolites and lipids. The periportal region was mainly associated with oxidative metabolism, while the pericentral region was linked to detoxification, lipogenesis, glycolysis, fatty acid synthesis, ketogenesis, and glycogen synthesis. Certain DAGs and FAs were specifically zonated to pericentral zones. Additionally, there was a disparity in FA zonation distribution between female and male liver tissues. Importantly, the study revealed that morphological homogeneous hepatocytes could be metabolically heterogeneous along the porto-central axis, clustering into distinct metabolic subpopulations.
Another study by Yang et al. [77] investigated spatial metabolic and lipidomic differentiation across different anatomical segments of the liver. Multiomic analyses (proteomic, lipidomic, and metabolomic) on tissues from eight liver segments created a segmentation atlas. Proteomic data indicated that the left lobe was primarily associated with energy metabolism, while the right lobe was linked to small molecule metabolism, including processes for toxic substances. Lipidomic data showed substantial differences between the left and right lobes, with variations in lipid groups among the eight segments. Although this study was not performed at single-cell resolution, it provided insights into the metabolic work distribution across different liver lobes and segments, integrating proteomic data to offer a comprehensive picture.
When metabolic disorders occur in the liver of patients who do not consume alcohol, it can lead to NAFLD or MASLD. In this condition, the hepatic lipid accumulation exposes hepatocytes to high concentrations of specific fatty acids, causing lipotoxicity. A study using mass spectrometry imaging (MSI) on manually annotated steatotic and non-steatotic regions of NAFLD tissues from 23 obese patients revealed spatial lipid distribution co-registered to histology images, creating spatial lipid signatures [78]. The study found lipid composition heterogeneity between steatotic and non-steatotic areas, identifying over 80 lipid species differentiating the two regions. Network enrichment analysis pinpointed metabolic pathways: PtdIns and arachidonic acid metabolism were linked to non-steatotic regions, while LDL and VLDL metabolism were associated with steatotic regions. This spatial lipid profiling indicated a metabolic transition from non-steatotic to steatotic status, highlighting the comprehensiveness of spatial metabolomic and lipidomic analyses coupled with protein network data to improve the understanding of NAFLD.
As NAFLD progresses, dead hepatocytes and the increasingly fibrotic hepatic microenvironment lead to cirrhosis. The low number of viable liver cells in cirrhotic tissues complicates metabolomics studies, explaining the limited research on metabolic heterogeneity in cirrhosis. However, one study on acute decompensation of cirrhosis found interpatient lipidomic heterogeneity [79]. Serum untargeted lipidomics in a cohort of 826 cirrhotic patients with acute decompensation (646 with acute-on-chronic liver failure and 180 without), and 29 healthy controls, identified 223 annotated lipids. Sphingomyelins distinguished decompensated and compensated cirrhosis, while cholesteryl esters differentiated decompensated cirrhotic patients with and without acute-on-chronic liver failure, revealing lipidomic heterogeneity in decompensated cirrhotic patients.
Many more studies have investigated metabolic heterogeneity in HCC tissues compared to cirrhotic tissues. For example, the Yang et al. study [77] performed multiomic analyses on 39 HCC tissues, stratifying them into three subtypes (C1-C3) based on their proteome. The C3 subtype had a higher abundance of lipids, including oleic acid, palmitic acid, arachidonic acid, and linoleic acid, demonstrating interpatient lipidomic heterogeneity potentially linked to proteomic differences. However, this study did not explore the proteomic and metabolomic/lipidomic relationship in depth, nor did it associate heterogeneity with clinicopathologic features.
Another study conducted untargeted metabolomic and lipidomic analyses and RNA-sequencing on nine pairs of HCC and corresponding non-tumorous liver tissues, identifying 3508 annotated lipids [39]. It observed mild interpatient heterogeneity in the lipidome and found enriched ceramide and reduced TAGs and sphingosine in HCC compared to non-tumorous tissues. LPCAT1 was identified as to be significantly upregulated in HCC and validated to play an oncogenic role via EGFR signaling activation. These studies, using various technology platforms, have addressed lipid metabolism heterogeneity at different stages of HCC development (Table 2).
Table 2. Summary of studies in the heterogeneity of lipid metabolism in liver and different developmental stages of HCC of MASLD-etiology.
To explore the plasticity and dynamic changes in response to metabolic perturbation upon exposure to fatty acid in the development of NAFLD, Shih et al. exposed HepG2 cells to fatty acids 13C16-palmitic acid and 3C16-palmitoleic acids and performed lipidomic analyses. They found differential lipidome between the exposures to the two fatty acids [80]. For palmitic acid-treated cells, there was a shift in lipid metabolic pathways towards ceramide, palmitoyl carnitine and LPA, to resolve the high concentrations of palmitic acid. For palmitoleic acid-treated cells, there was mainly a shift of metabolic pathway for generation of glycerolipids. This demonstrates the metabolic plasticity of the cells when exposed to different fatty acids may lead to lipid overloading in the heterogeneous development of NAFLD.
Beyond NAFLD, the plasticity of lipid metabolism in HCC cells has also been studied. It was discovered that knocking down NANOG activated the expression of multiple fatty acid synthesis genes (SCD1, FASN, and ACLY) [68]. Using gas chromatography/mass spectrometry (GC/MS) for lipid profiling, the ratio of C18:1/C16:1 FAs increased upon NANOG knockdown [68]. This suggests that NANOG inhibits fatty acid elongation or promotes fatty acid oxidation, indicating that NANOG encourages the use of fatty acids as an alternative energy source for cancer cell survival. Moreover, NANOG knockdown increased the expression of cytochrome c oxidase subunit 6A (Cox6a2), an enzyme supporting oxidative phosphorylation (OXPHOS), which facilitates the conversion of AMP/ADP to ATP [68]. This indicates that NANOG promotes metabolic reprogramming by enhancing fatty acid oxidation and inhibiting OXPHOS, thereby shifting the energy source from glucose to lipids. This study highlights how NANOG orchestrates cellular metabolic pathways to enable lipid metabolic plasticity in HCC.
Plasticity refers to the flexibility and ability of a cell to switch between different phenotypic states [9,81]. This includes processes such as (de)differentiation, transitions between epithelial and mesenchymal states, and changes in dependency on various metabolites [81]. In HCC, most studies have focused on EMT plasticity, with less emphasis on differentiation and metabolic plasticity, particularly in relation to lipids and lipid metabolism. In this section, we will explore the roles of lipids in regulating these different types of cellular plasticity in HCC.
Studies have demonstrated the roles of specific lipids and genes involved in lipid metabolism in promoting EMT in HCC. For example, butyrate-containing structured lipids (STLs) have been found to promote EMT in the rat "resistant hepatocyte" HCC model [82]. This model involves initial genotoxic insult using chemicals like DEN to induce pro-tumorigenic hepatocytes, followed by partial hepatectomy as regenerative stimuli, and administration of mitochondria-inhibitory reagents like 2-acetylaminofluorene (2-AAF) to selectively inhibit the proliferation of non-initiated hepatocytes, favoring the growth of preneoplastic foci and hyperplastic nodules, ultimately leading to HCC [83]. Using this model, researchers found that butyrate-containing STL treatment suppressed the expression of EMT markers SNAIL1 and vimentin, indicating EMT blockade [82]. This was associated with reduced expression of RACGAP1 and RAC1. Further in vitro experiments showed that siRNA-mediated knockdown of RAC1 reduced Snail1 and N-cadherin levels, implicating RAC1 signaling in the regulation of EMT by butyrate-containing lipids in HCC [82].
In addition to specific lipid moieties, the overexpression of the intracellular lipid transporter, high-density lipoprotein binding protein (HDLBP), induced by cholesterol synthesis, promotes EMT marker expression by binding to and stabilizing BRAF, driving HCC metastasis [84]. Another gene related to cholesterol metabolism, SOAT1, which is overexpressed in HCC, increases lipid droplet and cholesterol accumulation in HCC cells, promoting EMT [85].
Furthermore, serine/threonine-protein kinase 25 (STK25), which is upregulated in HCC and positively correlated with lipid synthesis-related genes ACC1 and ACLY, was found to promote EMT in HCC cells [86]. Knockdown of STK25 reduced lipid droplet formation, decreased lipid synthesis-related gene expression, and altered EMT marker expression, including increased E-cadherin and decreased N-cadherin, vimentin, and Snail proteins, indicative of EMT loss [86]. These findings highlight the intricate roles of lipid metabolism and related genes in regulating EMT in HCC.
In cancer, differentiated cells can exhibit plasticity by dedifferentiation to acquire stemness and become cancer stem cells [9]. To explore differentiation plasticity and its connection to lipid metabolism in HCC, we reviewed literature related to cancer stemness. Several lipid metabolism-related genes have been found to promote cancer stemness. For instance, SLC35C2, a solute carrier protein overexpressed in HCC and associated with poorer prognosis, upregulates various lipid metabolism-related genes, stimulating lipogenesis and lipid reprogramming, leading to lipid accumulation and enhanced cancer stemness [87]. Similarly, OCTN2, also overexpressed in HCC and linked to poorer prognosis, promotes cancer stemness properties such as sphere formation, tumorigenicity, and EpCAM expression by increasing fatty acid oxidation and oxidative phosphorylation [88]. In another study on HCC subclones derived from an AKT and NRAS-driven HCC mouse model, elevated expression of genes related to lipid uptake, synthesis, and oxidation was associated with distinct cancer stemness-related gene expression in the NHRI-80B4 subclone, which exhibited strong sphere-forming ability, indicating greater cancer stemness [89]. Interestingly, both endogenous and exogenous lipids were required to maintain this sphere-forming ability, underscoring the role of lipids in sustaining cancer stemness [89]. These findings highlight the significant role of lipid metabolism and related genes in regulating differentiation plasticity and maintaining cancer stemness in HCC.
Metabolic plasticity refers to the ability of cells to switch between different metabolic pathways to meet their energy demands [81]. This adaptability is often seen in cancer cells, enabling them to survive various nutritional deficiencies and stressful conditions, such as glucose and amino acid scarcity and hypoxia. Unlike differentiation and EMT plasticity, few studies focus on specific lipid moieties or lipid metabolism-related genes that regulate metabolic plasticity in HCC. However, some research highlights the use of lipids as alternative energy sources in HCC.
For example, a study on a tumorigenic derivative of the mouse HCC Hepa1-6 cell line demonstrated the cell line's ability to utilize fatty acids as an alternative energy source under glucose-deficient conditions [90]. Another study identified a specific lipid-related metabolite, sapienate fatty acid, which can serve as a surrogate lipid for cellular membranes, allowing plasticity in fatty acid desaturation pathways in HCC cells [91]. Typically, palmitate is desaturated into palmitoleate by SCD for membrane construction. Interestingly, cancer cell lines can be stratified into SCD-dependent, partially dependent, and independent types [91]. In SCD-independent cell lines, palmitate is desaturated into sapienate by fatty acid desaturase 2 (FADS2) and used as an alternative lipid source for membrane formation [91]. In SCD-dependent cell lines, sapienate supplementation or FADS2 overexpression could reverse the proliferation suppression caused by SCD inhibition [91]. This indicates that the ability to use sapienate for membrane biosynthesis provides plasticity in fatty acid desaturation, allowing cells to switch between using SCD and FADS for palmitate desaturation into palmitoleate and sapienate, respectively.
These findings demonstrate the role of FAs and certain lipid metabolites in enabling metabolic plasticity in HCC cells. However, further studies are needed to understand the specific roles of particular lipid moieties and lipid metabolism-related enzymes in modulating metabolic plasticity in HCC.
The pathogenesis of HCC is closely linked to chronic liver inflammation [92], often resulting from viral hepatitis [93], alcohol liver disease or MASLD [94,95]. The immune system's response to liver injury and regeneration is complex, influencing both tumor initiation and progression [96]. Therefore, understanding the immune landscape in HCC is essential for developing effective therapies and improving patient outcomes.
The liver serves as a unique immunological organ, balancing immune tolerance and activation [97]. Disruption of this balance plays a crucial role in the development of HCC. Dysregulated immune responses can promote tumorigenesis, with T cells, natural killer (NK) cells, macrophages, and dendritic cells shaping HCC progression. T cell responses, particularly those involving CD8+ cytotoxic T lymphocytes (CTLs), are integral to anti-tumor immunity [98]. However, chronic inflammation and persistent antigen exposure in the liver often lead to T cell exhaustion, marked by the upregulation of inhibitory receptors such as programmed cell death protein 1 (PD-1), T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), T cell immunoreceptor with Ig and ITIM domains (TIGIT), lymphocyte activation gene 3 (LAG-3), and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) [99–104]. Engagement of these receptors with their ligands, such as PD-L1 and B7 family proteins, results in the downregulation of T cell activation and effector functions, impairing anti-tumor responses and the efficacy of immune checkpoint inhibitor (ICI) treatments [105]. This immune dysfunction allows HCC cells to evade immune detection and contributes to tumor growth.
Tumor-associated macrophages (TAMs) can have either pro- or anti-tumor functions depending on their polarization. In many cases, TAMs in the HCC microenvironment adopt a pro-tumoral M2 phenotype, promoting tumor growth, angiogenesis, and immune evasion through the release of cytokines and chemokines, including interleukin (IL)-8, IL-10, VEGFA, FGF2, and C-C motif chemokine ligand (CCL)2 [106–110].
Chronic inflammation caused by viral infections, particularly hepatitis B virus (HBV) and hepatitis C virus (HCV), is a well-established risk factor for HCC. The continuous cycle of liver damage and repair activates various inflammatory pathways, including the nuclear factor-kappa B (NF-κB) and IL-6 signaling pathways, which contribute to hepatocyte proliferation and genomic instability [111–118]. Cytokines and chemokines released during chronic inflammation are key pro-inflammatory players in HCC. For example, tumor necrosis factor-alpha (TNF-α) induces NF-κB pathways that promote cell survival and proliferation, while IL-6 activates the STAT3 signaling pathway, associated with hepatocyte proliferation and anti-apoptotic effects [112,117]. Additionally, chemokines such as CCL2 and CCL5 facilitate the recruitment of immune cells to the liver, exacerbating inflammation and creating a microenvironment favorable for tumor development [110,119–122].
Recognition of the immune system's role in HCC has led to the use of immunotherapy as a treatment modality. Increased PD-L1 expression in some HCC tumors is associated with aggressive disease and poorer survival [123,124]. ICIs, such as anti-PD-1 and anti-CTLA4 antibodies, aim to reinvigorate the exhausted T cells and restore anti-tumor immunity, showing promise in various cancers, including HCC [125,126]. Additionally, LAG-3, another co-inhibitory receptor expressed in tumor-infiltrating lymphocytes in HCC, also plays a significant role in modulating anti-tumor immunity. LAG-3 has been shown to synergize with PD-1 in promoting HCC tumor progression [127]. Blocking or deleting LAG-3 reduces tumor growth in HCC transplant models and can enhance the effects of PD-1 inhibition [123]. Combining ICIs with other treatments, such as transarterial chemoembolization and targeted therapy, is an area of active research aimed at improving response rates and survival outcomes [128].
Lipid metabolic reprogramming in immune cells has emerged as a critical feature in various cancers, including HCC. To adapt to the nutrient-depleted and hypoxic microenvironment, immune cells rewire lipid metabolism pathways. This includes enhancing lipid or glucose uptake, lipid accumulation, fatty acid oxidation (FAO), and oxidative phosphorylation (OXPHOS) in mitochondria through the regulation of lipid-related gene expression and translation (Figure 5).
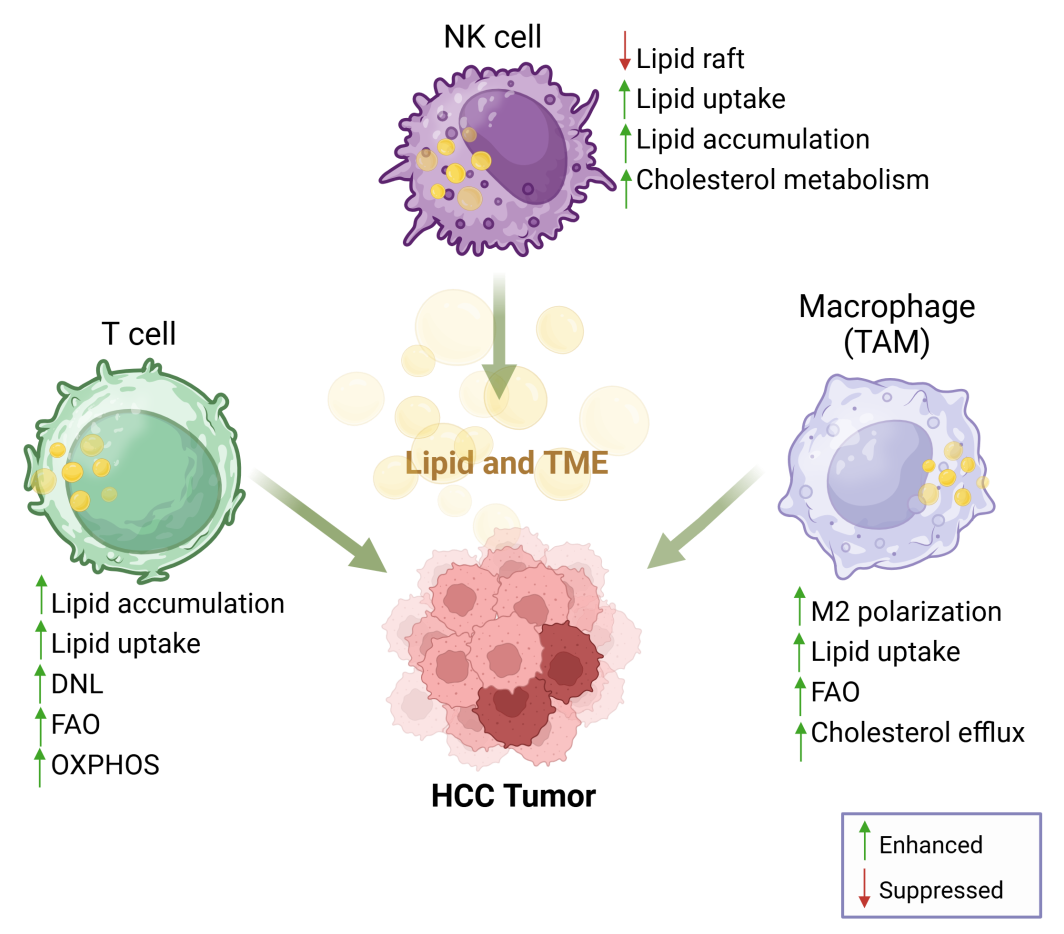
Figure 5. Reprogramming lipid metabolism in immune cells. Lipid metabolism and reprogramming play significant roles in tumorigenesis, lipid/cholesterol homeostasis, and serve as a crucial connection between innate immunity and adaptive immune responses. Figure was created by BioRender.com.
T cells play a crucial role in the adaptive immune response by distinguishing between 'self' and 'non-self' antigens and coordinating immune responses to defend against cancer. Extracellular and intracellular FAs, cholesterol, and cholesterol derivatives serve as sources of nutrients and energy for T cells, facilitating their proliferation and activation. The reprogrammed lipid metabolism in the tumor microenvironment (TME) reshapes the fate and function of T cells, potentially interfering with the efficacy of ICIs in HCC [129]. It is well-established that dietary lipids can affect lipid metabolite levels and their metabolism in HCC, further promoting tumor progression and metastasis [130]. Furthermore, MASLD or metabolic dysfunction-associated steatohepatitis (MASH) is a known risk factor for HCC [131]. The accumulation of excessive TAGs leads to hepatic steatosis, inflammation, MASH, fibrosis or cirrhosis and eventually tumorigenesis [132,133].
In preclinical MASLD-HCC models with liver-specific activation of MYC, such as those fed with methionine-choline deficient diet (MCD), choline-deficient and high-fat diet (CD-HFD), or induced with the DEN carcinogen, earlier HCC development was observed compared to normal diet controls [134]. This acceleration of tumorigenesis was accompanied by the selective loss of conventional intrahepatic CD4+ T cells, caused by exogenous dietary free FAs and lipid deposition. Abnormal oxidative phosphorylation, mitochondrial dysfunction, and apoptosis of anti-tumoral CD4+ T cells increased their intracellular reactive oxygen species (ROS) levels and promoted MASLD-HCC development [134,135]. Certain extracellular FAs affect the effector and memory responses of CD8+ T cells. In a CD-HFD-induced MASH-HCC mouse model, enrichment of auto-aggressive CXCR6-expressing CD8+ T cells exacerbated liver injuries and impeded the therapeutic effect of anti-PD-1. The upregulation of CXCR6 in CD8+ T cells, mediated by the IL-15/FOXO1 axis, made CD8+ T cells hypersensitive to extracellular FAs, triggering auto-aggressive killing in an MHC-class-I-independent manner [129,136]. Besides dietary lipids, gut microbiota-derived short-chain FAs, such as acetate, were found to suppress IL-17A expression in hepatic type 3 innate lymphoid cells and enhance anti-PD-1/anti-PD-L1 therapy in HCC [137].
While external lipids may impede anti-tumor immunity, intracellular lipids and their metabolites participate in mitochondrial FAO and oxidative phosphorylation (OXPHOS) in T cells to generate ATP [138]. FAO also produces acetyl-CoA for other cellular functions [138,139]. However, the mechanisms and interplay between T cells and FAO in the TME remain unclear. Under nutrient stress in the TME, lipid droplets (LDs) are deposited in CD8+ T cells via activation of acetyl-CoA carboxylase (ACC), inhibiting ATP-producing FAO in tumor-infiltrating T cells (TILs) and causing CD8+ T cell dysfunction [140]. In CTNNB1-mutated human HCC, FAO metabolic reprogramming in T cells is induced by peroxisome proliferator-activated receptor (PPAR) activation, promoting HCC development, while targeting FAO suppresses CTNNB1-mutated HCC [141]. Conversely, using agonists of PGC-1α/PPAR complexes enhances FAO in mitochondria, maintaining energy homeostasis in CD8+ T cells and allowing TIL activation and survival in fibrosarcoma [142].
Tumor-associated regulatory T cells (Tregs) are crucial regulators of immune evasion in the tumor microenvironment (TME). CD4+CD25+Foxp3+ Tregs have been found to be enriched in human HCC tumors and liver metastases, and their accumulation is associated with impaired tumor-specific T cell responses [143,144]. Importantly, while the metabolic shift from fatty acid oxidation (FAO) to glycolysis in T cells, which impairs cytotoxic T cell function, can be attributed to reduced de novo lipogenesis (DNL) due to the silencing of the SREBP1 pathway - a key transcription activator of lipogenic genes (e.g., SCD, ACC, FASN) - Tregs can survive in nutrient-depleted conditions and exert immunosuppressive effects on HCC [144].
Additionally, certain lipids can act as antigens presented by antigen-presenting cells to T cells, leading to T cell activation. Lipid antigens are organic molecules derived from lipids and are presented by atypical MHC class I proteins of the CD1 family (CD1a, CD1b, and CD1c) [145]. Lipid-reactive T cells are specialized in recognizing and responding to mycobacterial lipids and self-lipids presented by CD1, as opposed to peptide recognition by antigen receptors. These CD1-restricted T cells can sense changes in lipid composition in the physiological microenvironment during tumorigenesis [146]. Previous research revealed that CD1c-restricted T cells could recognize tumor-derived lipid antigens, such as methyl-lysophosphatidic acid (mLPA), in leukemia and exert tumor-killing effects in preclinical mouse models. CD1b-self-phospholipid-reactive T cells were reported to provide immune surveillance by responding to tumor-derived phospholipids in lymphoma mouse models. In HCC, a reduction in the CD1 family, particularly CD1d, was observed in AFP-exposed human dendritic cells (DCs), impairing the functions of natural killer T (NKT) cells. In summary, lipid metabolism in T cells represents a key interface between innate and adaptive immunity. Targeting lipid metabolism in T cells could be a potential therapeutic strategy in cancer treatment.
Natural Killer (NK) cells are cytotoxic lymphocytes that play a crucial role in the innate immune system. They can rapidly clear infected or cancerous cells by secreting lytic granules and enhancing immune responses without prior antigen exposure [147]. In patients with HBV or HCV infections, NK cells activate early and aid in viral clearance [148]. Studies have shown that the number and proportion of peripheral blood NK cells decreased in HCC patients. Further research indicates that lipid metabolism impacts NK cell signaling and functions by providing energy, inducing lipotoxicity, forming lipid rafts on cell membranes, and regulating immune checkpoints.
FAO is a vital energy source for optimal NK cell responses against viruses or cancers. Cytokine-associated activation of NK cells promotes both glycolysis and oxidative phosphorylation (OXPHOS). Activated NK cells enhance the transport of long-chain fatty acids into mitochondria for β-oxidation, maintaining their antitumor responses via upregulation of CPT1A in melanoma [147]. However, transcriptional reprogramming of NK cells to increase fatty acid metabolism through PPARγ upregulation suppresses interferon-γ (IFN-γ) production, impairing NK cell cytotoxic function in fatty acid-rich environments [149].
In high-fat diet (HFD)-induced obesity murine models, genes associated with lipid metabolism, including FABPs and low-density lipoprotein receptor (LDLR), were significantly upregulated compared to lean controls. PPARα/ẟ-driven lipid accumulation in NK cells led to dysfunction, marked by reduced IFN-γ secretion [150]. Mechanistically, exogenous fatty acid supplementation to NK cells impeded downstream mTORC1 activation, inhibiting their effector responses [150,151]. Conversely, NK cell depletion (Nfil3–/–) attenuated MASH development in MCD- and CD-HFD-fed mice by suppressing pro-inflammatory cytokines such as IFN-γ, IL-1β, CCL4/5, and granulocyte-macrophage colony-stimulating factor [152].
Conversely, a previous study revealed that high serum cholesterol suppressed HCC initiation and progression in LDLR knockout mice [153]. NK cells isolated from these mice, fed with a high-cholesterol diet, showed increased expression of stimulatory receptors NKG2D and NCR1, along with improved cell proliferation and survival. Further investigation indicated enhanced lipid raft formation in the plasma membrane of NK cells, which facilitated NKG2D and NCR1 localization and promoted NK cell-mediated signaling [153]. However, HFD-induced hyperlipidemia also impaired NK cell responses by reducing chromatin accessibility and stabilizing H3K27 deacetylation of genes encoding effector molecules such as IFN-γ, perforin, and granzyme B, thereby limiting anti-tumoral immunity in HCC [154].
In summary, lipid metabolism significantly influences NK cell function in HCC, potentially affecting disease progression and outcomes. However, further research is needed to fully understand these mechanisms and to develop effective therapeutic strategies.
Macrophages are vital components of the innate immune system and are distributed throughout the body. They primarily exist in two activated subpopulations: pro-inflammatory M1-like macrophages and anti-inflammatory M2-like macrophages [155]. In the context of cancer, M2-like tumor-associated macrophages (TAMs) are the predominant population within the HCC microenvironment, influencing HCC development and the effectiveness of ICI therapy [156,157]. Lipid metabolism plays a crucial role in modulating macrophage function and signaling, regulating their polarization to either the M1 or M2 phenotype, thereby impacting immune responses and the pathogenesis of various diseases, including cancer [158,159].
For example, DNL is enhanced in inflammatory macrophages upon activation of toll-like receptors (TLRs). During inflammation, increased glucose uptake promotes mitochondrial citrate production, elevating cytosolic acetyl-CoA levels to stimulate fatty acid synthesis [160]. The upregulation of the pentose phosphate pathway (PPP) and SREBP transcription activity further supports fatty acid synthesis and inflammatory cytokine production in macrophages [161]. In contrast, IL-4, a cytokine known for inducing M2 polarization, promotes exogenous triglyceride (TAG) uptake and lysosomal lipolysis, facilitating fatty acid oxidation (FAO), which is crucial for M2 activation [162].
Beyond fatty acid metabolism, dysregulated cholesterol metabolism in macrophages is also implicated in the pathogenesis of cardiovascular diseases, chronic liver diseases, and cancer [163,164]. Excessive cholesterol can be converted into cholesterol esters for storage or exported to the liver or tumor via high-density lipoprotein (HDL) through reverse cholesterol transport, fueling tumorigenesis [164,165]. Our previous study demonstrated that elevated cholesterol biosynthesis, driven by p90 ribosomal S6 kinase 2 mutations, sensitized the therapeutic effect of sorafenib in HBV-HCC patients [166]. Additionally, macrophage-derived cholesterol was found to regulate the nuclear localization of the androgen receptor, contributing to resistance to enzalutamide in prostate cancer [167].
Liver-resident macrophages, known as Kupffer cells (KCs), play a significant role in liver physiology, pathology, and oncology [168]. KCs can differentiate into mature macrophages, serving as key players in the immune response by secreting cytokines and chemokines and presenting antigens to T cells [147,160]. KCs also absorb and store lipids from the diet and from the lipolysis of white adipose tissue, leading to lipotoxicity and inflammation, which are considered precursors to malignancy [156,169]. In a high-fat diet (HFD)-induced metabolic dysfunction-associated steatotic liver disease (MASLD) murine model, KCs underwent morphological changes with lipid droplet accumulation, accompanied by aberrant lipid synthesis and trafficking [170]. This lipid metabolism reprogramming enhanced the secretion of pro-inflammatory cytokines and chemokines by KCs and promoted the recruitment of CD4+ T cells [170]. In summary, lipid metabolism and its reprogramming disrupt the lipid homeostasis of KCs, providing a critical link between innate and adaptive immune responses, which may contribute to tumorigenesis and progression.
Alterations in lipid profiles can significantly influence immune behavior, thereby affecting tumor surveillance, growth, and response to therapies in HCC. Although lipid metabolic reprogramming in HCC and immune cells has been discussed separately, their close proximity within TME enables crosstalk between HCC lipid metabolism and immune responses.
For instance, tumor-derived lipids can modulate the functions of immune cells, such as T cells and macrophages, potentially enhancing their ability to detect and eliminate tumor cells [171]. Inhibiting SCD1, which suppresses fatty acid desaturation, has been shown to inhibit tumor growth in tumor-bearing mice. This anti-tumor effect was abolished upon CD8+ T cell depletion, implicating a role for inflammatory pathways in SCD1-modulated tumor formation [172]. Additionally, a recent study revealed that FABP5-positive, lipid-loaded macrophages use tumor-derived long-chain unsaturated fatty acids to contribute to an immune-suppressive TME by activating the PPARγ pathway in HCC [173].
Lipid mediators like prostaglandins, synthesized by HCC cells, as discussed above, can promote tumor growth and invasiveness while enhancing immune evasion by recruiting Tregs and suppressing effector T cell function [67]. For instance, PGE2 has been reported to inhibit T cell proliferation through PGE2-induced indoleamine 2,3-dioxygenase (IDO) expression in DCs at high DC:T cell ratios, while at low DC:T ratios, PGE2 induces T-cell stimulatory functions in DCs [174]. Furthermore, prostaglandin receptor EP2 or EP4-dependent PGE2 signaling increases Foxp3 expression, driving the development of immunosuppressive CD4+CD25+ Treg cells in non-small cell lung carcinoma [175]. PGE2 from mouse renal carcinoma cells inhibited antitumor cytotoxic T lymphocytes (CTLs) by blocking IFNγ-dependent upregulation of ICAM-1, essential for the initial priming of naïve CD8+ T cells [176]. Through the PGE2 pathway, GM-CSF promotes Th9 cell differentiation and inhibits the differentiation of induced Treg cells [177]. PGE2 also promotes the expression of immune checkpoint inhibitors PD-1 and TIM-3 in T cells, leading to immunotherapy resistance [178]. Although prostaglandin synthesis is downstream from membrane phospholipid catabolism, these findings demonstrate the potential crosstalk between HCC lipid metabolism and anti-tumor immunity in the TME.
Targeting lipid metabolism offers a promising strategy for HCC treatment. By inhibiting lipogenic pathways or altering lipid profiles, it is possible to disrupt the metabolic support that tumors need for growth, metastasis and survival. For example, SCD1 overexpression in HCC supports tumor stemness and is linked to sorafenib resistance, making this enzyme a potential biomarker and therapeutic target for HCC treatment [179].
Lipid metabolites also hold potential as valuable diagnostics biomarkers in patient serum. Identifying specific lipid profiles associated with various stages of HCC could enhance early detection and monitoring of disease progression. Using ultra-high-performance liquid chromatography-Q Exactive mass spectrometry (UHPLC-Q-Exactive-MS) in the plasma of HCC and liver cirrhosis (LC) patients, significant variations were found in FAs, bile acids, and GPs [180]. Specifically, a dramatic increase in phosphatidylethanolamine and TAG was noted in HCC cases. These lipid metabolites showed better performance in HCC diagnosis compared with the traditional biomarker alpha-fetoprotein (AFP). Although further validation is needed, the identification and utilization of lipid biomarkers in HCC could lead to more effective treatment and better patient outcomes.
Moreover, targeting lipid metabolism through a personalized medicine approach can optimize treatment for individual patients. For example, CTNNB1-mutated HCC relies more on FAO for energy [181]. A recent multiomics study classified HCC patients into three subtypes (F1-3) based on their FAO profile: F1 subtype, with the lowest FAO activity and enriched with TP53 mutation, and the F3 subtype, with the highest FAO activity and more CTNNB1 mutations [182]. The study found that the F1 subtype responded better to sorafenib and ICIs like anti-PD-1/PD-L1 therapy, while the F3 subtype showed better responses to transarterial chemoembolization [182]. These findings demonstrate that tailoring therapies based on specific lipid metabolic profiles can help clinicians develop more effective, patient-centered treatment plans that enhance efficacy while minimizing adverse effects.
The significant role of lipid metabolism in modulating immune responses in HCC presents a promising strategy to enhance therapeutic outcomes through anti-tumor immunity. Inhibitors of lipogenic enzymes, such as FASN, have demonstrated potential in preclinical models by enhancing anti-tumor immunity and improving treatment efficacy [183,184].
Manipulating lipid metabolism and signaling in immune cells can also promote anti-tumor immunity in HCC therapy. For instance, ceramides, important signaling lipids, have shown anti-proliferative and pro-apoptotic effects on HCC cells [185]. Exogenous C6-ceramide has been found to delay liver tumor formation in mice by enhancing the activity of antigen-specific CD8+ T cells and inhibiting the M2-like phenotype of tumor-associated macrophages (TAMs) [186]. This demonstrated lipid metabolites may have dual effects on both tumor and immune cells. Targeting cellular ceramide synthesis could be a future therapeutic direction, offering an alternative to the current exogenous nanoliposomal form of ceramide.
Although ICIs targeting PD-1, PD-L1 and CTLA-4 have been approved for treating advanced HCC, the response rate remains around 30% [187]. Combining lipid metabolism modulators with ICIs may enhance immunotherapy efficacy, potentially overcoming lipid-mediated immune evasion resistance [184]. For example, TPST-1120, an oral inhibitor of PPARo that regulates fatty acid metabolism, has shown promise in early clinical trials. In a Phase I study (NCT03829436), TPST-1120 demonstrated tolerability and efficacy, with some patients achieving stable disease or objective responses, even including those previously resistant to anti-PD-1 therapy [188]. Another example is avasimibe, an inhibitor of the cholesterol esterification enzyme acetyl-CoA acetyltransferase 1 (ACAT1) that has shown enhanced anti-tumoral efficacy when combined with anti-PD-1 in melanoma [189]. ACAT1 inhibition increases cholesterol levels in T-cell plasma membranes, enhancing CD8+ T cell clustering and maturation. Combination of avasimibe with chimeric antigen receptor-T cell therapy has also shown improved outcome in HBV and other solid tumors, supporting the potential of lipid metabolism inhibition in conjunction with ICI.
Immune checkpoints can also affect metabolism. During CD4+ T cell activation via PD-1 ligation, glucose intake and glycolysis are inhibited, while fatty acid oxidation (FAO) is enhanced via CPT1A and ATGL [190,191]. However, non-activated T cells exhibit impaired glycolysis without promoting CTLA-4-regulated FAO [190]. FDA-approved ICIs, including ipilimumab (anti-CTLA-4), nivolumab/pembrolizumab (anti-PD-1), and atezolizumab (anti-PD-L1), not only can block the immune checkpoint, but also inhibit FAO [144]. Further research is needed to understand how immune checkpoints regulate tumor immunity and to explore the targeting of immunity-associated lipid reprogramming for novel combination therapies.
In conclusion, the future direction of cancer therapy leveraging lipid metabolism involves a multifaceted approach, including therapeutics, diagnostic biomarker development, elucidation of immune vulnerabilities, enhancement of immune responses, and personalized treatment strategies. While monotherapy with ICIs against HCC is suboptimal, combined treatment with inhibitors targeting tumor or immune lipid metabolism offers a promising strategy for improving HCC treatment. Identifying and incorporating plasma lipid-based biomarkers for early HCC detection could complement these interventions, holding significant potential for improving cancer management and patient outcomes.
The authors declare no competing interests.
This project was supported in part by grants from the Research Grants Council of Hong Kong Theme-based Research Scheme (T12-716/22R), Innovation and Technology Commission grant to State Key Laboratory of Liver Research (ITC PD/17-9), National Natural Science Foundation of China (No. 82203234 and 82394451), and University Development Fund and Seed Fund for Basic Research (2201101565) and Translational and Applied Research (2303101596) of The University of Hong Kong. I Ng is Loke Yew Professor in Pathology.
T.C.Y., Y.M.T., V.X.Z., H.M. and I.O.N drafted the manuscript. All authors reviewed and approved the final manuscript.
The following abbreviations are used in this manuscript:
| 1. | Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. [Google Scholar] [CrossRef] |
| 2. | Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J Hepatol. 2020;73(1):202-209. [Google Scholar] [CrossRef] |
| 3. | Cao LQ, Xie Y, Fleishman JS, Liu X, Chen ZS. Hepatocellular carcinoma and lipid metabolism: Novel targets and therapeutic strategies. Cancer Lett. 2024;597:217061. [Google Scholar] [CrossRef] |
| 4. | Cheng K, Cai N, Zhu J, Yang X, Liang H, Zhang W. Tumor-associated macrophages in liver cancer: From mechanisms to therapy. Cancer Commun (Lond). 2022;42(11):1112-1140. [Google Scholar] [CrossRef] |
| 5. | Wang W, Wang C, Xu H, Gao Y. Aldehyde Dehydrogenase, Liver Disease and Cancer. Int J Biol Sci. 2020;16(6):921-934. [Google Scholar] [CrossRef] |
| 6. | Wu K, Lin F. Lipid Metabolism as a Potential Target of Liver Cancer. J Hepatocell Carcinoma. 2024;11:327-346. [Google Scholar] [CrossRef] |
| 7. | Xu K, Xia P, Gongye X, Zhang X, Ma S, Chen Z, et al. A novel lncRNA RP11-386G11.10 reprograms lipid metabolism to promote hepatocellular carcinoma progression. Mol Metab. 2022;63:101540. [Google Scholar] [CrossRef] |
| 8. | Xu K, Xia P, Chen X, Ma W, Yuan Y. ncRNA-mediated fatty acid metabolism reprogramming in HCC. Trends Endocrinol Metab. 2023;34(5):278-291. [Google Scholar] [CrossRef] |
| 9. | Huch M, Dolle L. The plastic cellular states of liver cells: Are EpCAM and Lgr5 fit for purpose? Hepatology. 2016;64(2):652-662. [Google Scholar] [CrossRef] |
| 10. | Xu K, Xia P, Liu P, Zhang X. A six lipid metabolism related gene signature for predicting the prognosis of hepatocellular carcinoma. Sci Rep. 2022;12(1):20781. [Google Scholar] [CrossRef] |
| 11. | Fahy E, Subramaniam S, Brown HA, Glass CK, Merrill AH Jr., Murphy RC, et al. A comprehensive classification system for lipids. J Lipid Res. 2005;46(5):839-861. [Google Scholar] [CrossRef] |
| 12. | Nguyen P, Leray V, Diez M, Serisier S, Le Bloc'h J, Siliart B, et al. Liver lipid metabolism. J Anim Physiol Anim Nutr (Berl). 2008;92(3):272-283. [Google Scholar] [CrossRef] |
| 13. | Bond LM, Miyazaki M, O’Neill LM, Ding F, Ntambi JM. Chapter 6 - Fatty Acid Desaturation and Elongation in Mammals. In: Ridgway ND, McLeod RS, editors. Biochemistry of Lipids, Lipoproteins and Membranes (Sixth Edition). Boston: Elsevier; 2016. p. 185–208. [CrossRef] |
| 14. | Schulz H. Beta oxidation of fatty acids. BBA-Mol Cell Biol L. 1991;1081(2):109-120. [Google Scholar] [CrossRef] |
| 15. | Dyall SC, Balas L, Bazan NG, Brenna JT, Chiang N, da Costa Souza F, et al. Polyunsaturated fatty acids and fatty acid-derived lipid mediators: Recent advances in the understanding of their biosynthesis, structures, and functions. Prog Lipid Res. 2022;86:101165. [Google Scholar] [CrossRef] |
| 16. | Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res. 2004;43(2):134-176. [Google Scholar] [CrossRef] |
| 17. | Takeuchi K, Reue K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am J Physiol Endocrinol Metab. 2009;296(6):E1195-1209. [Google Scholar] [CrossRef] |
| 18. | McFie PJ, Patel A, Stone SJ. The monoacylglycerol acyltransferase pathway contributes to triacylglycerol synthesis in HepG2 cells. Sci Rep. 2022;12(1):4943. [Google Scholar] [CrossRef] |
| 19. | Zadoorian A, Du X, Yang H. Lipid droplet biogenesis and functions in health and disease. Nat Rev Endocrinol. 2023;19(8):443-459. [Google Scholar] [CrossRef] |
| 20. | Gluchowski NL, Becuwe M, Walther TC, Farese RV Jr. Lipid droplets and liver disease: from basic biology to clinical implications. Nat Rev Gastroenterol Hepatol. 2017;14(6):343-355. [Google Scholar] [CrossRef] |
| 21. | Lian J, Nelson R, Lehner R. Carboxylesterases in lipid metabolism: from mouse to human. Prot Cell. 2018;9(2):178-195. [Google Scholar] [CrossRef] |
| 22. | Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, et al. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell. 2015;163(2):340-353. [Google Scholar] [CrossRef] |
| 23. | Lee H, Horbath A, Kondiparthi L, Meena JK, Lei G, Dasgupta S, et al. Cell cycle arrest induces lipid droplet formation and confers ferroptosis resistance. Nat Commun. 2024;15(1):79. [Google Scholar] [CrossRef] |
| 24. | Ackerman D, Tumanov S, Qiu B, Michalopoulou E, Spata M, Azzam A, et al. Triglycerides Promote Lipid Homeostasis during Hypoxic Stress by Balancing Fatty Acid Saturation. Cell Rep. 2018;24(10):2596-605.e5. [Google Scholar] [CrossRef] |
| 25. | Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr., Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100(6):3077-3082. [Google Scholar] [CrossRef] |
| 26. | Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, Zoncu R, et al. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev Cell. 2017;42(1):9-21.e5. [Google Scholar] [CrossRef] |
| 27. | Wang X, Devaiah SP, Zhang W, Welti R. Signaling functions of phosphatidic acid. Prog. Lipid Res. 2006;45(3):250-278. [Google Scholar] [CrossRef] |
| 28. | Cooke M, Kazanietz MG. Overarching roles of diacylglycerol signaling in cancer development and antitumor immunity. Sci Signal. 2022;15(729):eabo0264. [Google Scholar] [CrossRef] |
| 29. | Brose N, Betz A, Wegmeyer H. Divergent and convergent signaling by the diacylglycerol second messenger pathway in mammals. Current Opinion in Neurobiology. 2004;14(3):328-340. [Google Scholar] [CrossRef] |
| 30. | Carrasco S, Mérida I. Diacylglycerol, when simplicity becomes complex. Trends Biochem Sci. 2007;32(1):27-36. [Google Scholar] [CrossRef] |
| 31. | Wang B, Tontonoz P. Phospholipid Remodeling in Physiology and Disease. Annu Rev Physiol. 2019;81:165-188. [Google Scholar] [CrossRef] |
| 32. | Cooper AD. Hepatic uptake of chylomicron remnants. J Lipid Res. 1997;38(11):2173-2192. [Google Scholar] [CrossRef] |
| 33. | Luo J, Yang H, Song B-L. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. 2020;21(4):225-245. [Google Scholar] [CrossRef] |
| 34. | Cao D, Liu H. Dysregulated cholesterol regulatory genes in hepatocellular carcinoma. Eur J Med Res. 2023;28(1):580. [Google Scholar] [CrossRef] |
| 35. | Yeagle PL. Cholesterol and the cell membrane. Biochim Biophys Acta. 1985;822(3–4):267-287. [Google Scholar] [CrossRef] |
| 36. | Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1(1):31-39. [Google Scholar] [CrossRef] |
| 37. | Sangineto M, Villani R, Cavallone F, Romano A, Loizzi D, Serviddio G. Lipid Metabolism in Development and Progression of Hepatocellular Carcinoma. Cancers (Basel). 2020;12(6):1419. [Google Scholar] [CrossRef] |
| 38. | Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. 2020;122(1):4-22. [Google Scholar] [CrossRef] |
| 39. | Liu Q, Zhang X, Qi J, Tian X, Dovjak E, Zhang J, et al. Comprehensive profiling of lipid metabolic reprogramming expands precision medicine for HCC. Hepatology. 2024;81(4):1164-1180. [Google Scholar] [CrossRef] |
| 40. | Zhang J, Zhang Z, Wu Z, Wang Y, Zhang Z, Xia L. The switch triggering the invasion process: Lipid metabolism in the metastasis of hepatocellular carcinoma. Chin Med J (Engl). 2024;137(11):1271-1284. [Google Scholar] [CrossRef] |
| 41. | Paul B, Lewinska M, Andersen JB. Lipid alterations in chronic liver disease and liver cancer. JHEP Rep. 2022;4(6):100479. [Google Scholar] [CrossRef] |
| 42. | Li Z, Liao X, Hu Y, Li M, Tang M, Zhang S, et al. SLC27A4-mediated selective uptake of mono-unsaturated fatty acids promotes ferroptosis defense in hepatocellular carcinoma. Free Radic Biol Med. 2023;201:41-54. [Google Scholar] [CrossRef] |
| 43. | Liu HH, Xu Y, Li CJ, Hsu SJ, Lin XH, Zhang R, et al. An SCD1-dependent mechanoresponsive pathway promotes HCC invasion and metastasis through lipid metabolic reprogramming. Mol Ther. 2022;30(7):2554-2567. [Google Scholar] [CrossRef] |
| 44. | Cheng Y, He J, Zuo B, He Y. Role of lipid metabolism in hepatocellular carcinoma. Discov Oncol. 2024;15(1):206. [Google Scholar] [CrossRef] |
| 45. | Jia J, Che L, Cigliano A, Wang X, Peitta G, Tao J, et al. Pivotal Role of Fatty Acid Synthase in c-MYC Driven Hepatocarcinogenesis. Int J Mol Sci. 2020;21(22). [Google Scholar] [CrossRef] |
| 46. | Grube J, Woitok MM, Mohs A, Erschfeld S, Lynen C, Trautwein C, et al. ACSL4-dependent ferroptosis does not represent a tumor-suppressive mechanism but ACSL4 rather promotes liver cancer progression. Cell Death Dis. 2022;13(8):704. [Google Scholar] [CrossRef] |
| 47. | Cheng L, Zhu Y, Han H, Zhang Q, Cui K, Shen H, et al. MicroRNA-148a deficiency promotes hepatic lipid metabolism and hepatocarcinogenesis in mice. Cell Death Dis. 2017;8(7):e2916. [Google Scholar] [CrossRef] |
| 48. | Fu R, Xue W, Liang J, Li X, Zheng J, Wang L, et al. SOAT1 regulates cholesterol metabolism to induce EMT in hepatocellular carcinoma. Cell Death Dis. 2024;15(5):325. [Google Scholar] [CrossRef] |
| 49. | Yang Z, Qin W, Chen Y, Yuan B, Song X, Wang B, et al. Cholesterol inhibits hepatocellular carcinoma invasion and metastasis by promoting CD44 localization in lipid rafts. Cancer Lett. 2018;429:66-77. [Google Scholar] [CrossRef] |
| 50. | Carr BI, Giannelli G, Guerra V, Giannini EG, Farinati F, Rapaccini GL, et al. Plasma cholesterol and lipoprotein levels in relation to tumor aggressiveness and survival in HCC patients. Int J Biol Marker. 2018;33(4):423-431. [Google Scholar] [CrossRef] |
| 51. | Jiang SS, Weng DS, Jiang L, Zhang YJ, Pan K, Pan QZ, et al. The clinical significance of preoperative serum cholesterol and high-density lipoprotein-cholesterol levels in hepatocellular carcinoma. J Cancer. 2016;7(6):626-632. [Google Scholar] [CrossRef] |
| 52. | Zhao Z, Zhong L, He K, Qiu C, Li Z, Zhao L, et al. Cholesterol attenuated the progression of DEN-induced hepatocellular carcinoma via inhibiting SCAP mediated fatty acid de novo synthesis. Biochem Biophys Res Commun. 2019;509(4):855-861. [Google Scholar] [CrossRef] |
| 53. | Li Y, Li T, Jin Y, Shen J. Dgat2 reduces hepatocellular carcinoma malignancy via downregulation of cell cycle-related gene expression. Biomed Pharmacother. 2019;115:108950. [Google Scholar] [CrossRef] |
| 54. | Wang H, Bell M, Sreenivasan U, Sreenevasan U, Hu H, Liu J, et al. Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J Biol Chem. 2011;286(18):15707-15715. [Google Scholar] [CrossRef] |
| 55. | Mass-Sanchez PB, Krizanac M, Štancl P, Leopold M, Engel KM, Buhl EM, et al. Perilipin 5 deletion protects against nonalcoholic fatty liver disease and hepatocellular carcinoma by modulating lipid metabolism and inflammatory responses. Cell Death Discov. 2024;10(1):94. [Google Scholar] [CrossRef] |
| 56. | Zhang Y, Liang X, Lian Q, Liu L, Zhang B, Dong Z, et al. Transcriptional analysis of the expression and prognostic value of lipid droplet-localized proteins in hepatocellular carcinoma. BMC Cancer. 2023;23(1):677. [Google Scholar] [CrossRef] |
| 57. | Meng Y, Guo D, Lin L, Zhao H, Xu W, Luo S, et al. Glycolytic enzyme PFKL governs lipolysis by promoting lipid droplet-mitochondria tethering to enhance β-oxidation and tumor cell proliferation. Nat Metab. 2024;6(6):1092-1107. [Google Scholar] [CrossRef] |
| 58. | Liu M, Yu X, Lin L, Deng J, Wang K, Xia Y, et al. ATGL promotes the proliferation of hepatocellular carcinoma cells via the p-AKT signaling pathway. J Biochem Mol Toxicol. 2019;33(11):e22391. [Google Scholar] [CrossRef] |
| 59. | Liu X, Liang Y, Song R, Yang G, Han J, Lan Y, et al. Long non-coding RNA NEAT1-modulated abnormal lipolysis via ATGL drives hepatocellular carcinoma proliferation. Mol Cancer. 2018;17(1):90. [Google Scholar] [CrossRef] |
| 60. | Hatano M, Akiyama Y, Shimada S, Yagi K, Akahoshi K, Itoh M, et al. Loss of KDM6B epigenetically confers resistance to lipotoxicity in nonalcoholic fatty liver disease-related HCC. Hepatol Commun. 2023;7(10):e0277. [Google Scholar] [CrossRef] |
| 61. | Zhang J, Liu Z, Lian Z, Liao R, Chen Y, Qin Y, et al. Monoacylglycerol Lipase: A Novel Potential Therapeutic Target and Prognostic Indicator for Hepatocellular Carcinoma. Sci Rep. 2016;6:35784. [Google Scholar] [CrossRef] |
| 62. | Di Leo L, Vegliante R, Ciccarone F, Salvatori I, Scimeca M, Bonanno E, et al. Forcing ATGL expression in hepatocarcinoma cells imposes glycolytic rewiring through PPAR-α/p300-mediated acetylation of p53. Oncogene. 2019;38(11):1860-1875. [Google Scholar] [CrossRef] |
| 63. | Lin T, Zhang E, Lin Z, Peng L. Comprehensive Analysis of LPCATs Highlights the Prognostic and Immunological Values of LPCAT1/4 in Hepatocellular Carcinoma. Int J Gen Med. 2021;14:9117-9130. [Google Scholar] [CrossRef] |
| 64. | Morita Y, Sakaguchi T, Ikegami K, Goto-Inoue N, Hayasaka T, Hang VT, et al. Lysophosphatidylcholine acyltransferase 1 altered phospholipid composition and regulated hepatoma progression. J Hepatol. 2013;59(2):292-299. [Google Scholar] [CrossRef] |
| 65. | VanSant-Webb C, Low HK, Kuramoto J, Stanley CE, Qiang H, Su AY, et al. Phospholipid isotope tracing suggests β-catenin-driven suppression of phosphatidylcholine metabolism in hepatocellular carcinoma. Biochim Biophys Acta Mol Cell Biol Lipids. 2024;1869(6):159514. [Google Scholar] [CrossRef] |
| 66. | Tian Y, Jellinek MJ, Mehta K, Seok SM, Kuo SH, Lu W, et al. Membrane phospholipid remodeling modulates nonalcoholic steatohepatitis progression by regulating mitochondrial homeostasis. Hepatology. 2024;79(4):882-897. [Google Scholar] [CrossRef] |
| 67. | Chen C, Guan J, Gu X, Chu Q, Zhu H. Prostaglandin E2 and Receptors: Insight Into Tumorigenesis, Tumor Progression, and Treatment of Hepatocellular Carcinoma. Front Cell Dev Biol. 2022;10:834859. [Google Scholar] [CrossRef] |
| 68. | Chen CL, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, et al. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab. 2016;23(1):206-219. [Google Scholar] [CrossRef] |
| 69. | Li G, Li X, Mahmud I, Ysaguirre J, Fekry B, Wang S, et al. Interfering with lipid metabolism through targeting CES1 sensitizes hepatocellular carcinoma for chemotherapy. JCI Insight. 2023;8(2):e163624. [Google Scholar] [CrossRef] |
| 70. | Lin M, Lv D, Zheng Y, Wu M, Xu C, Zhang Q, et al. Downregulation of CPT2 promotes tumorigenesis and chemoresistance to cisplatin in hepatocellular carcinoma. Onco Targets Ther. 2018;11:3101-3110. [Google Scholar] [CrossRef] |
| 71. | Raeisi M, Hassanbeigi L, Khalili F, Kharrati-Shishavan H, Yousefi M, Mehdizadeh A. Stearoyl-CoA desaturase 1 as a therapeutic target for cancer: a focus on hepatocellular carcinoma. Mol Biol Rep. 2022;49(9):8871-8882. [Google Scholar] [CrossRef] |
| 72. | Bansal S, Berk M, Alkhouri N, Partrick DA, Fung JJ, Feldstein A. Stearoyl-CoA desaturase plays an important role in proliferation and chemoresistance in human hepatocellular carcinoma. J Surg Res. 2014;186(1):29-38. [Google Scholar] [CrossRef] |
| 73. | Krohler T, Kessler SM, Hosseini K, List M, Barghash A, Patial S, et al. The mRNA-binding Protein TTP/ZFP36 in Hepatocarcinogenesis and Hepatocellular Carcinoma. Cancers (Basel). 2019;11(11):1754. [Google Scholar] [CrossRef] |
| 74. | Wang X, Zhang L, Dong B. Molecular mechanisms in MASLD/MASH-related HCC. Hepatology. 2024. [Google Scholar] [CrossRef] |
| 75. | Seubnooch P, Montani M, Tsouka S, Claude E, Rafiqi U, Perren A, et al. Characterisation of hepatic lipid signature distributed across the liver zonation using mass spectrometry imaging. JHEP Rep. 2023;5(6):100725. [Google Scholar] [CrossRef] |
| 76. | Tian H, Rajbhandari P, Tarolli J, Decker AM, Neelakantan TV, Angerer T, et al. Multimodal mass spectrometry imaging identifies cell-type-specific metabolic and lipidomic variation in the mammalian liver. Dev Cell. 2024;59(7):869-881 e6. [Google Scholar] [CrossRef] |
| 77. | Yang S, Zheng L, Li L, Zhang J, Wang J, Zhao H, et al. Integrative multiomics analysis identifies molecular subtypes and potential targets of hepatocellular carcinoma. Clin Transl Med. 2024;14(6):e1727. [Google Scholar] [CrossRef] |
| 78. | Scupakova K, Soons Z, Ertaylan G, Pierzchalski KA, Eijkel GB, Ellis SR, et al. Spatial Systems Lipidomics Reveals Nonalcoholic Fatty Liver Disease Heterogeneity. Anal Chem. 2018;90(8):5130-5138. [Google Scholar] [CrossRef] |
| 79. | Claria J, Curto A, Moreau R, Colsch B, Lopez-Vicario C, Lozano JJ, et al. Untargeted lipidomics uncovers lipid signatures that distinguish severe from moderate forms of acutely decompensated cirrhosis. J Hepatol. 2021;75(5):1116-1127. [Google Scholar] [CrossRef] |
| 80. | Shih LM, Tang HY, Lynn KS, Huang CY, Ho HY, Cheng ML. Stable Isotope-Labeled Lipidomics to Unravel the Heterogeneous Development Lipotoxicity. Molecules. 2018;23(11):2862. [Google Scholar] [CrossRef] |
| 81. | Tsui YM, Chan LK, Ng IO. Cancer stemness in hepatocellular carcinoma: mechanisms and translational potential. Br J Cancer. 2020;122(10):1428-4140. [Google Scholar] [CrossRef] |
| 82. | de Conti A, Tryndyak V, Heidor R, Jimenez L, Moreno FS, Beland FA, et al. Butyrate-containing structured lipids inhibit RAC1 and epithelial-to-mesenchymal transition markers: a chemopreventive mechanism against hepatocarcinogenesis. J Nutr Biochem. 2020;86:108496. [Google Scholar] [CrossRef] |
| 83. | Romualdo GR, Leroy K, Costa CJS, Prata GB, Vanderborght B, da Silva TC, et al. In Vivo and In Vitro Models of Hepatocellular Carcinoma: Current Strategies for Translational Modeling. Cancers (Basel). 2021;13(21):1-57. [Google Scholar] [CrossRef] |
| 84. | Yuan J, Lv T, Yang J, Wu Z, Yan L, Yang J, et al. The lipid transporter HDLBP promotes hepatocellular carcinoma metastasis through BRAF-dependent epithelial-mesenchymal transition. Cancer Lett. 2022;549:215921. [Google Scholar] [CrossRef] |
| 85. | Fu R, Xue W, Liang J, Li X, Zheng J, Wang L, et al. SOAT1 regulates cholesterol metabolism to induce EMT in hepatocellular carcinoma. Cell Death Dis. 2024;15(5):325. [Google Scholar] [CrossRef] |
| 86. | Zhang Y, Xu J, Qiu Z, Guan Y, Zhang X, Zhang X, et al. STK25 enhances hepatocellular carcinoma progression through the STRN/AMPK/ACC1 pathway. Cancer Cell Int. 2022;22(1):4. [Google Scholar] [CrossRef] |
| 87. | Qi C, Cao B, Gong Z, Zhang W, Yang P, Qin H, et al. SLC35C2 promotes stemness and progression in hepatocellular carcinoma by activating lipogenesis. Cell Signal. 2025;127:111589. [Google Scholar] [CrossRef] |
| 88. | Yang T, Liang N, Zhang J, Bai Y, Li Y, Zhao Z, et al. OCTN2 enhances PGC-1alpha-mediated fatty acid oxidation and OXPHOS to support stemness in hepatocellular carcinoma. Metabolism. 2023;147:155628. [Google Scholar] [CrossRef] |
| 89. | Cheng YH, Ko YC, Ku HJ, Huang CC, Yao YC, Liao YT, et al. Novel Paired Cell Lines for the Study of Lipid Metabolism and Cancer Stemness of Hepatocellular Carcinoma. Front Cell Dev Biol. 2022;10:821224. [Google Scholar] [CrossRef] |
| 90. | Cassim S, Raymond VA, Dehbidi-Assadzadeh L, Lapierre P, Bilodeau M. Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle. 2018;17(7):903-916. [Google Scholar] [CrossRef] |
| 91. | Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A, et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature. 2019;566(7744):403-406. [Google Scholar] [CrossRef] |
| 92. | Czaja AJ. Hepatic inflammation and progressive liver fibrosis in chronic liver disease. World J Gastroenterol. 2014;20(10):2515-2532. [Google Scholar] [CrossRef] |
| 93. | El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142(6):1264-73 e1. [Google Scholar] [CrossRef] |
| 94. | Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol. 2012;56(6):1384-1391. [Google Scholar] [CrossRef] |
| 95. | Koh MY, Gagea M, Sargis T, Lemos R Jr., Grandjean G, Charbono A, et al. A new HIF-1alpha/RANTES-driven pathway to hepatocellular carcinoma mediated by germline haploinsufficiency of SART1/HAF in mice. Hepatology. 2016;63(5):1576-1591. [Google Scholar] [CrossRef] |
| 96. | Thorgersen EB, Barratt-Due A, Haugaa H, Harboe M, Pischke SE, Nilsson PH, et al. The Role of Complement in Liver Injury, Regeneration, and Transplantation. Hepatology. 2019;70(2):725-736. [Google Scholar] [CrossRef] |
| 97. | Robinson MW, Harmon C, O'Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. 2016;13(3):267-276. [Google Scholar] [CrossRef] |
| 98. | Giles JR, Globig AM, Kaech SM, Wherry EJ. CD8(+) T cells in the cancer-immunity cycle. Immunity. 2023;56(10):2231-2253. [Google Scholar] [CrossRef] |
| 99. | Alison MR, Lin WR. Macrophages come on tap for liver fibrosis therapy. Hepatology. 2018;68(3):1194-1196. [Google Scholar] [CrossRef] |
| 100. | Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. 2017;66(5):1037-1046. [Google Scholar] [CrossRef] |
| 101. | Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, et al. A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell. 2016;166(6):1500-11 e9. [Google Scholar] [CrossRef] |
| 102. | Topper MJ, Vaz M, Marrone KA, Brahmer JR, Baylin SB. The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol. 2020;17(2):75-90. [Google Scholar] [CrossRef] |
| 103. | Xie C, Duffy AG, Mabry-Hrones D, Wood B, Levy E, Krishnasamy V, et al. Tremelimumab in Combination With Microwave Ablation in Patients With Refractory Biliary Tract Cancer. Hepatology. 2019;69(5):2048-2060. [Google Scholar] [CrossRef] |
| 104. | Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. 2020;20(3):173-185. [Google Scholar] [CrossRef] |
| 105. | Cheung TT, Wai-Hung Ho D, Lyu SX, Zhang Q, Tsui YM, Ching-Yun Yu T, et al. Multimodal Integrative Genomics and Pathology Analyses in Neoadjuvant Nivolumab Treatment for Intermediate and Locally Advanced Hepatocellular Carcinoma. Liver Cancer. 2024;13(1):70-88. [Google Scholar] [CrossRef] |
| 106. | Geginat J, Larghi P, Paroni M, Nizzoli G, Penatti A, Pagani M, et al. The light and the dark sides of Interleukin-10 in immune-mediated diseases and cancer. Cytokine Growth Factor Rev. 2016;30:87-93. [Google Scholar] [CrossRef] |
| 107. | Shields CWt, Evans MA, Wang LL, Baugh N, Iyer S, Wu D, et al. Cellular backpacks for macrophage immunotherapy. Sci Adv. 2020;6(18):eaaz6579. [Google Scholar] [CrossRef] |
| 108. | Im JH, Buzzelli JN, Jones K, Franchini F, Gordon-Weeks A, Markelc B, et al. FGF2 alters macrophage polarization, tumour immunity and growth and can be targeted during radiotherapy. Nat Commun. 2020;11(1):4064. [Google Scholar] [CrossRef] |
| 109. | Zhang X, Yu C, Zhao S, Wang M, Shang L, Zhou J, et al. The role of tumor-associated macrophages in hepatocellular carcinoma progression: A narrative review. Cancer Med. 2023;12(24):22109-22129. [Google Scholar] [CrossRef] |
| 110. | Zhou C, Weng J, Liu C, Liu S, Hu Z, Xie X, et al. Disruption of SLFN11 Deficiency-Induced CCL2 Signaling and Macrophage M2 Polarization Potentiates Anti-PD-1 Therapy Efficacy in Hepatocellular Carcinoma. Gastroenterology. 2023;164(7):1261-1278. [Google Scholar] [CrossRef] |
| 111. | Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. [Google Scholar] [CrossRef] |
| 112. | Nakagawa H. ER Stress Cooperates with Hypernutrition to Trigger TNF-Dependent Spontaneous HCC Development. Cancer Cell. 2014;26(3):331-343. [Google Scholar] [CrossRef] |
| 113. | Zhao C, Li H, Lin HJ, Yang S, Lin J, Liang G. Feedback Activation of STAT3 as a Cancer Drug-Resistance Mechanism. Trends Pharmacol Sci. 2016;37(1):47-61. [Google Scholar] [CrossRef] |
| 114. | Hirano T. IL-6 in inflammation, autoimmunity and cancer. Int Immunol. 2021;33(3):127-148. [Google Scholar] [CrossRef] |
| 115. | Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798-809. [Google Scholar] [CrossRef] |
| 116. | Ding YF, Wu ZH, Wei YJ, Shu L, Peng YR. Hepatic inflammation-fibrosis-cancer axis in the rat hepatocellular carcinoma induced by diethylnitrosamine. J Cancer Res Clin Oncol. 2017;143(5):821-834. [Google Scholar] [CrossRef] |
| 117. | Shao YY, Lin H, Li YS, Lee YH, Chen HM, Cheng AL, et al. High plasma interleukin-6 levels associated with poor prognosis of patients with advanced hepatocellular carcinoma. Jpn J Clin Oncol. 2017;47(10):949-953. [Google Scholar] [CrossRef] |
| 118. | He G. Identification of Liver Cancer Progenitors Whose Malignant Progression Depends on Autocrine IL-6 Signaling. Cell. 2013;155(2):384-396. [Google Scholar] [CrossRef] |
| 119. | Xu H, Zhao J, Li J, Zhu Z, Cui Z, Liu R, et al. Cancer associated fibroblast-derived CCL5 promotes hepatocellular carcinoma metastasis through activating HIF1alpha/ZEB1 axis. Cell Death Dis. 2022;13(5):478. [Google Scholar] [CrossRef] |
| 120. | Xia Y, Zhou L, Yang HC, Yu CW. Chemokine CCL5 immune subtypes of human liver cancer with prognostic significance. Int Immunopharmacol. 2022;113(Pt A):109372. [Google Scholar] [CrossRef] |
| 121. | Mohs A, Kuttkat N, Reissing J, Zimmermann HW, Sonntag R, Proudfoot A, et al. Functional role of CCL5/RANTES for HCC progression during chronic liver disease. J Hepatol. 2017;66(4):743-753. [Google Scholar] [CrossRef] |
| 122. | Liu Y, Yang Y, Wang T, Wang L, Wang X, Li T, et al. Long non-coding RNA CCAL promotes hepatocellular carcinoma progression by regulating AP-2alpha and Wnt/beta-catenin pathway. Int J Biol Macromol. 2017;109:424-434. [Google Scholar] [CrossRef] |
| 123. | Yarchoan M, Xing D, Luan L, Xu H, Sharma RB, Popovic A, et al. Characterization of the Immune Microenvironment in Hepatocellular Carcinoma. Clin Cancer Res. 2017;23(23):7333-7339. [Google Scholar] [CrossRef] |
| 124. | Gao Q, Wang XY, Qiu SJ, Yamato I, Sho M, Nakajima Y, et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009;15(3):971-979. [Google Scholar] [CrossRef] |
| 125. | Kaseb AO, Vence L, Blando J, Yadav SS, Ikoma N, Pestana RC, et al. Immunologic Correlates of Pathologic Complete Response to Preoperative Immunotherapy in Hepatocellular Carcinoma. Cancer Immunol Res. 2019;7(9):1390-1395. [Google Scholar] [CrossRef] |
| 126. | Onuma AE, Zhang H, Huang H, Williams TM, Noonan A, Tsung A. Immune Checkpoint Inhibitors in Hepatocellular Cancer: Current Understanding on Mechanisms of Resistance and Biomarkers of Response to Treatment. Gene Expr. 2020;20(1):53-65. [Google Scholar] [CrossRef] |
| 127. | Schnell A, Bod L, Madi A, Kuchroo VK. The yin and yang of co-inhibitory receptors: toward anti-tumor immunity without autoimmunity. Cell Res. 2020;30(4):285-299. [Google Scholar] [CrossRef] |
| 128. | Shigeta K, Datta M, Hato T, Kitahara S, Chen IX, Matsui A, et al. Dual Programmed Death Receptor-1 and Vascular Endothelial Growth Factor Receptor-2 Blockade Promotes Vascular Normalization and Enhances Antitumor Immune Responses in Hepatocellular Carcinoma. Hepatology. 2020;71(4):1247-1261. [Google Scholar] [CrossRef] |
| 129. | Pfister D, Nunez NG, Pinyol R, Govaere O, Pinter M, Szydlowska M, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592(7854):450-456. [Google Scholar] [CrossRef] |
| 130. | Jin HR, Wang J, Wang ZJ, Xi MJ, Xia BH, Deng K, et al. Lipid metabolic reprogramming in tumor microenvironment: from mechanisms to therapeutics. J Hematol Oncol. 2023;16(1):103. [Google Scholar] [CrossRef] |
| 131. | Younossi ZM, Loomba R, Rinella ME, Bugianesi E, Marchesini G, Neuschwander-Tetri BA, et al. Current and Future Therapeutic Regimens for Non-alcoholic Fatty Liver Disease (NAFLD) and Non-alcoholic Steatohepatitis (NASH). Hepatology. 2018;68(1):361-371. [Google Scholar] [CrossRef] |
| 132. | Ebrahimi F, Hagstrom H, Sun J, Bergman D, Shang Y, Yang W, et al. Familial coaggregation of MASLD with hepatocellular carcinoma and adverse liver outcomes: Nationwide multigenerational cohort study. J Hepatol. 2023;79(6):1374-84. [Google Scholar] [CrossRef] |
| 133. | Riazi K, Azhari H, Charette JH, Underwood FE, King JA, Afshar EE, et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol. 2022;7(9):851-861. [Google Scholar] [CrossRef] |
| 134. | Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531(7593):253-257. [Google Scholar] [CrossRef] |
| 135. | Weiskirchen R, Tacke F. Immune surveillance of liver cancer in non-alcoholic fatty liver disease: excess lipids cause CD4 T-cells loss and promote hepatocellular carcinoma development. Hepatobiliary Surg Nutr. 2016;5(5):433-437. [Google Scholar] [CrossRef] |
| 136. | Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, Laschinger M, et al. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature. 2021;592(7854):444-449. [Google Scholar] [CrossRef] |
| 137. | Hu C, Xu B, Wang X, Wan WH, Lu J, Kong D, et al. Gut microbiota-derived short-chain fatty acids regulate group 3 innate lymphoid cells in HCC. Hepatology. 2023;77(1):48-64. [Google Scholar] [CrossRef] |
| 138. | Chen Q, Du J, Cui K, Fang W, Zhao Z, Chen Q, et al. Acetyl-CoA derived from hepatic mitochondrial fatty acid beta-oxidation aggravates inflammation by enhancing p65 acetylation. iScience. 2021;24(11):103244. [Google Scholar] [CrossRef] |
| 139. | Liu S, Lai J, Feng Y, Zhuo Y, Zhang H, Chen Y, et al. Acetyl-CoA carboxylase 1 depletion suppresses de novo fatty acid synthesis and mitochondrial beta-oxidation in castration-resistant prostate cancer cells. J Biol Chem. 2023;299(1):102720. [Google Scholar] [CrossRef] |
| 140. | Hunt EG, Hurst KE, Riesenberg BP, Kennedy AS, Gandy EJ, Andrews AM, et al. Acetyl-CoA carboxylase obstructs CD8(+) T cell lipid utilization in the tumor microenvironment. Cell Metab. 2024;36(5):969-983 e10. [Google Scholar] [CrossRef] |
| 141. | Senni N, Savall M, Cabrerizo Granados D, Alves-Guerra MC, Sartor C, Lagoutte I, et al. beta-catenin-activated hepatocellular carcinomas are addicted to fatty acids. Gut. 2019;68(2):322-334. [Google Scholar] [CrossRef] |
| 142. | Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8(+) T Cells and Facilitates Anti-PD-1 Therapy. Cancer Immunol Res. 2018;6(11):1375-87. [Google Scholar] [CrossRef] |
| 143. | Pedroza-Gonzalez A, Verhoef C, Ijzermans JNM, Peppelenbosch MP, Kwekkeboom J, Verheij J, et al. Activated tumor-infiltrating CD4+regulatory T cells restrain antitumor immunity in patients with primary or metastatic liver cancer. Hepatology. 2013;57(1):183-194. [Google Scholar] [CrossRef] |
| 144. | Hu B, Lin JZ, Yang XB, Sang XT. Aberrant lipid metabolism in hepatocellular carcinoma cells as well as immune microenvironment: A review. Cell Proliferat. 2020;53(3):e12772. [Google Scholar] [CrossRef] |
| 145. | Dowds CM, Kornell SC, Blumberg RS, Zeissig S. Lipid antigens in immunity. Biol. Chem. 2014;395(1):61-81. [Google Scholar] [CrossRef] |
| 146. | De Libero G, Mori L. Recognition of lipid antigens by T cells. Nat Rev Immunol. 2005;5(6):485-496. [Google Scholar] [CrossRef] |
| 147. | Sheppard S, Srpan K, Lin W, Lee M, Delconte RB, Owyong M, et al. Fatty acid oxidation fuels natural killer cell responses against infection and cancer. Proc Natl Acad Sci U S A. 2024;121(11):e2319254121. [Google Scholar] [CrossRef] |
| 148. | Sun C, Sun HY, Xiao WH, Zhang C, Tian ZG. Natural killer cell dysfunction in hepatocellular carcinoma and NK cell-based immunotherapy. Acta Pharmacol Sin. 2015;36(10):1191-1199. [Google Scholar] [CrossRef] |
| 149. | Kobayashi T, Lam PY, Jiang H, Bednarska K, Gloury R, Murigneux V, et al. Increased lipid metabolism impairs NK cell function and mediates adaptation to the lymphoma environment. Blood. 2020;136(26):3004-3017. [Google Scholar] [CrossRef] |
| 150. | Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol. 2018;19(12):1330-1340. [Google Scholar] [CrossRef] |
| 151. | Li Y, Zhang Y, Lu R, Dai M, Shen M, Zhang J, et al. Lipid metabolism changes in patients with severe COVID-19. Clin Chim Acta. 2021;517:66-73. [Google Scholar] [CrossRef] |
| 152. | Wang F, Zhang X, Liu W, Zhou Y, Wei W, Liu D, et al. Activated Natural Killer Cell Promotes Nonalcoholic Steatohepatitis Through Mediating JAK/STAT Pathway. Cell Mol Gastroenterol Hepatol. 2022;13(1):257-274. [Google Scholar] [CrossRef] |
| 153. | Qin WH, Yang ZS, Li M, Chen Y, Zhao XF, Qin YY, et al. High Serum Levels of Cholesterol Increase Antitumor Functions of Nature Killer Cells and Reduce Growth of Liver Tumors in Mice. Gastroenterology. 2020;158(6):1713-1727. [Google Scholar] [CrossRef] |
| 154. | Jiao D, Sun R, Ren X, Wang Y, Tian P, Wang Y, et al. Lipid accumulation-mediated histone hypoacetylation drives persistent NK cell dysfunction in anti-tumor immunity. Cell Rep. 2023;42(10):113211. [Google Scholar] [CrossRef] |
| 155. | Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877:173090. [Google Scholar] [CrossRef] |
| 156. | Xie Q, Zeng Y, Zhang X, Yu F. The significance of lipid metabolism reprogramming of tumor-associated macrophages in hepatocellular carcinoma. Cancer Immunol Immunother. 2024;73(9):171. [Google Scholar] [CrossRef] |
| 157. | Panda S, Arora A, Luthra K, Mohan A, Vikram NK, Kumar Gupta N, et al. Hyperglycemia modulates M1/M2 macrophage polarization in chronic diabetic patients with pulmonary tuberculosis infection. Immunobiology. 2024;229(2):152787. [Google Scholar] [CrossRef] |
| 158. | O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553-565. [Google Scholar] [CrossRef] |
| 159. | Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity. 2010;33(5):699-712. [Google Scholar] [CrossRef] |
| 160. | Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15(4):323-332. [Google Scholar] [CrossRef] |
| 161. | Yan J, Horng T. Lipid Metabolism in Regulation of Macrophage Functions. Trends Cell Biol. 2020;30(12):979-989. [Google Scholar] [CrossRef] |
| 162. | Huang SCC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15(9):846-855. [Google Scholar] [CrossRef] |
| 163. | Leleu D, Pilot T, Mangin L, Van Dongen K, Proukhnitzky L, Pais-De-Barros JP, et al. Impact of Liver X Receptor Inhibition on Macrophage Cholesterol Homeostasis and Inflammatory Response in Atherosclerosis. Atherosclerosis. 2024;395. [Google Scholar] [CrossRef] |
| 164. | Bendre S, Krawczynska N, Bhogale S, Weisser E, Han S, Hajyousif B, et al. The Loss of Cholesterol Efflux Pump ABCA1 Skews Macrophages Towards a Pro-tumor Phenotype and Facilitates Breast Cancer Progression. Cancer Immunol Res. 2024;12 10_Supplement:A048. [Google Scholar] [CrossRef] |
| 165. | Kim J, Kim JY, Byeon HE, Kim JW, Kim HA, Suh CH, et al. Inhibition of Toll-like Receptors Alters Macrophage Cholesterol Efflux and Foam Cell Formation. Int J Mol Sci. 2024;83:1436. [Google Scholar] [CrossRef] |
| 166. | Chan LK, Ho DW, Kam CS, Chiu EY, Lo IL, Yau DT, et al. RSK2-inactivating mutations potentiate MAPK signaling and support cholesterol metabolism in hepatocellular carcinoma. J Hepatol. 2021;74(2):360-371. [Google Scholar] [CrossRef] |
| 167. | El-Kenawi A, Dominguez-Viqueira W, Liu M, Awasthi S, Abraham-Miranda J, Keske A, et al. Macrophage-Derived Cholesterol Contributes to Therapeutic Resistance in Prostate Cancer. Cancer Res. 2021;81(21):5477-5490. [Google Scholar] [CrossRef] |
| 168. | Wu H, Zhong Z, Wang A, Yuan C, Ning K, Hu H, et al. LncRNA FTX represses the progression of non-alcoholic fatty liver disease to hepatocellular carcinoma via regulating the M1/M2 polarization of Kupffer cells. Cancer Cell Int. 2020;20:266. [Google Scholar] [CrossRef] |
| 169. | Wu K, Kryczek I, Chen L, Zou W, Welling TH. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 2009;69(20):8067-8075. [Google Scholar] [CrossRef] |
| 170. | Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol. 2012;57(1):141-149. [Google Scholar] [CrossRef] |
| 171. | Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell. 2017;169(4):570-586. [Google Scholar] [CrossRef] |
| 172. | Katoh Y, Yaguchi T, Kubo A, Iwata T, Morii K, Kato D, et al. Inhibition of stearoyl-CoA desaturase 1 (SCD1) enhances the antitumor T cell response through regulating beta-catenin signaling in cancer cells and ER stress in T cells and synergizes with anti-PD-1 antibody. J Immunother Cancer. 2022;10(7):e004616. [Google Scholar] [CrossRef] |
| 173. | Yang X, Deng B, Zhao W, Guo Y, Wan Y, Wu Z, et al. FABP5(+) lipid-loaded macrophages process tumour-derived unsaturated fatty acid signal to suppress T-cell antitumour immunity. J Hepatol. 2024;82(4):676-689. [Google Scholar] [CrossRef] |
| 174. | Shimabukuro-Vornhagen A, Liebig TM, Koslowsky T, Theurich S, von Bergwelt-Baildon MS. The ratio between dendritic cells and T cells determines whether prostaglandin E2 has a stimulatory or inhibitory effect. Cell Immunol. 2013;281(1):62-67. [Google Scholar] [CrossRef] |
| 175. | Sharma S, Yang SC, Zhu L, Reckamp K, Gardner B, Baratelli F, et al. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res. 2005;65(12):5211-5220. [Google Scholar] [CrossRef] |
| 176. | Basingab FS, Ahmadi M, Morgan DJ. IFNgamma-Dependent Interactions between ICAM-1 and LFA-1 Counteract Prostaglandin E2-Mediated Inhibition of Antitumor CTL Responses. Cancer Immunol Res. 2016;4(5):400-411. [Google Scholar] [CrossRef] |
| 177. | Kim IK, Koh CH, Jeon I, Shin KS, Kang TS, Bae EA, et al. GM-CSF Promotes Antitumor Immunity by Inducing Th9 Cell Responses. Cancer Immunol Res. 2019;7(3):498-509. [Google Scholar] [CrossRef] |
| 178. | Gorchs L, Fernandez Moro C, Bankhead P, Kern KP, Sadeak I, Meng Q, et al. Human Pancreatic Carcinoma-Associated Fibroblasts Promote Expression of Co-inhibitory Markers on CD4(+) and CD8(+) T-Cells. Front Immunol. 2019;10:847. [Google Scholar] [CrossRef] |
| 179. | Ma MKF, Lau EYT, Leung DHW, Lo J, Ho NPY, Cheng LKW, et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J Hepatol. 2017;67(5):979-990. [Google Scholar] [CrossRef] |
| 180. | Rashid MM, Varghese RS, Ding Y, Ressom HW. Biomarker Discovery for Hepatocellular Carcinoma in Patients with Liver Cirrhosis Using Untargeted Metabolomics and Lipidomics Studies. Metabolites. 2023;13(10):1047. [Google Scholar] [CrossRef] |
| 181. | Senni N, Savall M, Cabrerizo Granados D, Alves-Guerra MC, Sartor C, Lagoutte I, et al. β-catenin-activated hepatocellular carcinomas are addicted to fatty acids. Gut. 2019;68(2):322-34. [Google Scholar] [CrossRef] |
| 182. | Li B, Li Y, Zhou H, Xu Y, Cao Y, Cheng C, et al. Multiomics identifies metabolic subtypes based on fatty acid degradation allocating personalized treatment in hepatocellular carcinoma. Hepatology. 2024;79(2):289-306. [Google Scholar] [CrossRef] |
| 183. | Cuyas E, Pedarra S, Verdura S, Pardo MA, Espin Garcia R, Serrano-Hervas E, et al. Fatty acid synthase (FASN) is a tumor-cell-intrinsic metabolic checkpoint restricting T-cell immunity. Cell Death Discov. 2024;10(1):417. [Google Scholar] [CrossRef] |
| 184. | Wang H, Zhou Y, Xu H, Wang X, Zhang Y, Shang R, et al. Therapeutic efficacy of FASN inhibition in preclinical models of HCC. Hepatology. 2022;76(4):951-966. [Google Scholar] [CrossRef] |
| 185. | Tagaram HRS, DiVittore NA, Barth BM, Kaiser JM, Avella D, Kimchi ET, et al. Nanoliposomal ceramide prevents in vivo growth of hepatocellular carcinoma. Gut. 2011;60(5):695. [Google Scholar] [CrossRef] |
| 186. | Li G, Liu D, Kimchi ET, Kaifi JT, Qi X, Manjunath Y, et al. Nanoliposome C6-Ceramide Increases the Anti-tumor Immune Response and Slows Growth of Liver Tumors in Mice. Gastroenterology. 2018;154(4):1024-1036 e9. [Google Scholar] [CrossRef] |
| 187. | Sangro B, Sarobe P, Hervas-Stubbs S, Melero I. Advances in immunotherapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2021;18(8):525-543. [Google Scholar] [CrossRef] |
| 188. | Yarchoan M, Powderly JD, Bastos BR, Karasic TB, Crysler OV, Munster PN, et al. First-in-human Phase I Trial of TPST-1120, an Inhibitor of PPARalpha, as Monotherapy or in Combination with Nivolumab, in Patients with Advanced Solid Tumors. Cancer Res Commun. 2024;4(4):1100-1110. [Google Scholar] [CrossRef] |
| 189. | Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature. 2016;531(7596):651-5. [Google Scholar] [CrossRef] |
| 190. | Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692. [Google Scholar] [CrossRef] |
| 191. | Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, et al. Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell Metab. 2018;28(3):504-515 e7. [Google Scholar] [CrossRef] |

![]()
Copyright © 2025 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy




