Abstract
Cancer consists of heterogeneous cells, including cancer stem cells (CSCs), cancer cells, and tumor-associated cells, such as immune cells and vascular cells. Considering that these diverse cell types influence one another directly and indirectly through membrane proteins and secretion factors, such as exosomes and growth factors, the overall heterogeneity affects tumorigenicity and resistance to therapy. This review explores cancer heterogeneity, focusing on CSC heterogeneity, and discussed how the heterogeneity emerges by the intrinsic mechanism and the external factors and affects response to therapy. Additionally, as a potential therapeutic strategy to address this heterogeneity, I propose new Adeno-associated virus carrying a miRNA-dependent CSC eradication system that targets all types of CSCs with minimizing side effects.
Keywords
Glioblasoma (GBM), GBM-initiating cells (GICs), heterogeneity, stem cell factors, microRNA (miR), Adeno-Associated virus (AAV)
1. Introduction
Efforts to develop effective cancer treatments have revealed numerous aspects of the disease, including oncogenes, tumor suppressor genes, signaling pathways, CSCs, secretory factors (such as exosomes), and immune checkpoints [1]. Despite these advancements, successful cancer treatment remains challenging. One reason is that the complete cellular communication network involved in tumorigenesis has not been fully understood. By utilizing advanced technologies like single-cell RNA sequencing (scRNAseq analysis) and spatial transcriptomic analysis of tumors, it has become apparent that cancer consists of heterogeneous cell populations, including CSCs, cancer cells, and neighboring cells such as immune cells, blood vessels, and tissue-specific resident cells [2,3].
Furthermore, a tumor is likely to contain multiple types of CSCs that are generated during the process of tumorigenesis and by the reversion of cancer cells to CSCs induced by the microenvironment factors, such as Transforming growth factor β1 (TGFβ1) [4–6]. This indicates that each CSC in a certain cancer may have a different molecular profile. While CSCs generally possess strong tumorigenic abilities and give rise to various types of cancer cells, each individual CSC may exhibit different resistance to radiation and anticancer drugs, contributing to multidrug resistance in the tumor and likelihood of recurrence [7–11]. Therefore, gaining a comprehensive understanding of the mechanisms underlying CSC heterogeneity and plasticity throughout the entire cancer is crucial for the development of effective therapies.
Many researchers have extensively studied and characterized cancer stem cells (CSCs) and have identified potential therapeutic targets (Table 1). However, it is important to note that these targets also play a crucial role in maintaining the normal function of tissues and stem cells. For instance, CD133, a well-known marker for CSCs, is expressed in various normal adult stem cell types and is essential for the survival of photoreceptor cells [12–17]. Another membrane protein, CD44, found in CSCs, serves as a homing factor for immune cells and a cell-adhesion protein in keratinocytes [18,19]. Additionally, many other membrane proteins, such as CD24 [20,21], CD49f [21–23], CD15 [24,25] and LGR5 [26,27], are expressed in various types of CSC, while ABC transporters [28,29] and Aldehyde Dehydrogenase (ALDH) [30,31] act as detoxification factors in CSC as well as normal adult stem cells. Dihydroorotate Dehydrogenase (DHODH), a key factor in pyrimidine synthesis, is considered a potential therapeutic target for CSCs [32–38]. However, it is important to note that DHODH is also crucial for the proliferation of blast cells, which protect the body against infectious diseases and cancer [39]. Therefore, there is a concern about potential side effects if these CSC factors are targeted directly without a specific delivery method for CSCs.
Emerging evidence suggests that cancer cells acquire CSC status in the specific environment, such as the hypoxia [40]. Additionally, studies have shown that oligodendrocyte precursor cells (OPCs) and astrocytes (ASTs) can acquire multi-potency similar to neural stem cells (NSCs) under specific culture conditions [41,42]. Understanding which cancer cells revert to CSCs and uncovering the mechanisms of plasticity are crucial steps in developing novel therapeutic strategies.
This review discusses how cancer heterogeneity is generated and how it contributes to therapy resistance. Furthermore, we will explore potential methods for eradicating CSCs beyond the scope of CSC heterogeneity.
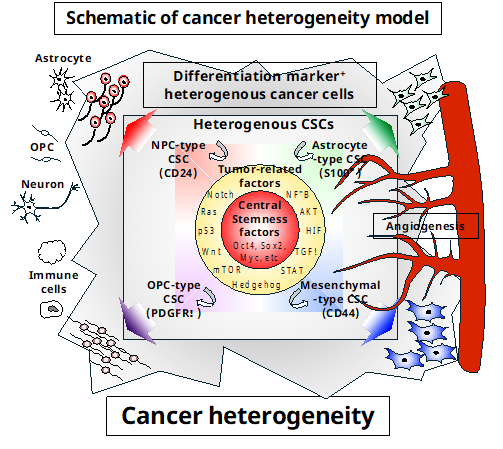
2. Cancer heterogeneity
Analysis of specimens using scRNAseq revealed that cancer consists of heterogeneous cells [43–45]. In the case of glioblastoma (GBM), for example, GBM stem-like cells (GSCs) commonly express central stemness factors, such as Oct4, Sox2, and Myc. The combined expression of tumor-related factors, including Nuclear factor-κB (NF-κB), RAC-Alpha Serine/Threonine-Protein Kinase (AKT), Hypoxia inducible factor (HIF), TGFβ, Signal Transducer and Activator of Transcription 3 (STAT3), Hedgehog, mechanistic Target of Rapamycin (mTOR), Wingless-Type MMTV Integration Site Family (Wnt), p53, Rat Sarcoma Viral Oncogene Homolog (Ras), and Notch, as well as cell lineage-specific genes (such as CD24 for neuronal precursor cells (NPCs), S100β for AST, Platelet-derived growth factor receptor α (PDGFRα) for oligodendrocyte precursor cells (OPCs), and CD44 for mesenchymal cells, MES), reveals the heterogeneity of GSC [46]. These heterogeneous GSCs communicate with one another and the tumor microenvironment through membrane proteins and secretion factors to promote maintenance, proliferation, and the production of cancer cell (Figure 1). Furthermore, heterogeneous GSCs in GBM were found to have amplifications in the copy number of Cyclin-dependent kinase 4 (CDK4), Epidermal growth factor receptor (EGFR), and PDGFRα loci as well as mutations in the Neurofibromin 1 (NF1) locus [43]. This demonstrates that tumors are more complex and heterogeneous than initially believed.
Abdelfattah et al. demonstrated that GBM consists of approximately 40% cancer cells, 45% myeloid cells, and 10% T cells, based on scRNAseq analysis of over 200,000 cells from 44 specimens [47]. The molecular characteristics of GBM further confirmed that each tumor contained a mixture of GSCs with NPC-like, OPC-like, AST-like, and MES-like features at different ratios. T cells were found to consist of eight clusters, including three CD8+ clusters, two CD4+ clusters, one naive T cell cluster, and two NK cell clusters. One of the CD4 clusters expressed regulatory T cell markers like FOXP3 and CTLA4, while expression of PD1 was low in NK and T cells across all samples, indicating an immunosuppressive environment in GBM [48]. Additionally, myeloid cells, the largest stromal compartment in GBM, comprised nine clusters, which did not correlate with the in vitro-defined macrophage states (M0, M1, or M2), but rather included both anti-tumorigenic and immunosuppressive macrophages. The study also found that microglia clusters had an anti-tumorigenic effect, whereas macrophage and MES-derived stem cell clusters were pro-tumorigenic.
Cytometry using Time-of-Flight (CyTOF) mass cytometry is another technology used for analyzing tumor heterogeneity. Wagner et al. prepared a single-cell atlas for tumors and immune cells in breast cancer using mass cytometry [49]. They discovered phenotypic abnormalities and phenotype dominance in breast cancer cells. Through immunohistochemical analysis, they also observed a relationship between PD-L1+ tumor-associated macrophages and exhausted T cells in high-grade tumors, revealing characteristics of ecosystems related to immunosuppression and poor prognosis. One limitation of mass cytometry analysis is the use of established antibodies (up to 40) in one experiment. Additionally, this technique cannot determine which cells are bona fide CSCs, as the membrane proteins specific to pan-CSCs have not yet been identified.
Spatial transcriptomics analysis is a latest technology. Yu et al. combined scRNAseq with multi-sector biopsies of gliomas to generate a geographical gene-expression dataset of the tumor, revealing the cellular status at the single cell level in the tumor and the surrounding microenvironment [50]. They found that each GBM contains four GSC subtypes: NPC-type, OPC-type, AST-type and MES-type, at different ratio. They also discovered that high interaction between OPC-type and MES-type, particularly in the hypoxia subtype, correlated with worse prognosis for the patients. Furthermore, Ravi et al. combined spatial transcriptomics with metabolomics and proteomics and showed that GBM is organized by spatial segregation of lineage states based on the locoregional tumor-host interdependence and the selection pressure of environmental stress [51].
Considering that these heterogeneous cells communicate with each other directly through cell membrane proteins and indirectly through secretion factors to maintain cancer cell homeostasis, this complex environment poses challenges for the development of effective therapeutics.
3. CSC as an origin of intratumor heterogeneity
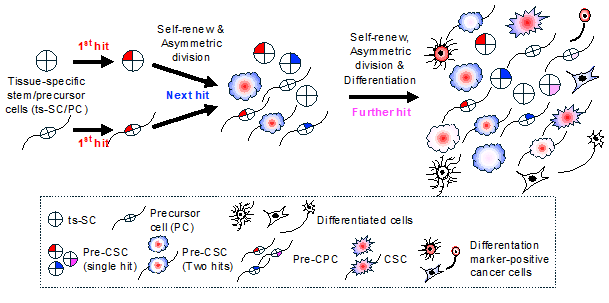
How is cancer heterogeneity generated? At the beginning of tumorigenesis, an initial genetic or epigenetic mutation occurs in the original cancer cell (Figure 2). If tissue specific stem cell (ts-SC) is the cell-of-origin of cancer, then the ts-SC with a genetic or epigenetic mutation (pre-CSC) acts as a seed of intratumor heterogeneity and can produce multiple pre-cancer precursor cell (pre-CPC) types. During proliferation, pre-CSCs may accumulate further random mutations over time, eventually transforming into various CSCs with different characteristics. This is one of the processes that result in the expansion of intratumor heterogeneity. Conversely, pre-CPCs are thought to either transform into a lineage-committed CPC, which forms a low-grade tumor, or become CSCs by acquiring both stemness and tumorigenic properties after accumulating additional hits [52,53]. These processes also increase intratumor heterogeneity.
Acquiring stemness associated with transformation can be reproduced in culture. For example, our study demonstrated that when p53 knockout (p53KO) OPCs overexpressed oncogenic H-RasL61 (where glycine at codon 61 is replaced with leucine), they acquired both tumorigenicity and stemness [53]. These H-RasL61 overexpressing p53KO OPCs (OPCL61) lost their original characteristics, such as the expression of Olig2 and Nkx2.2, but began to express neural stem cell (NSC) markers, including High mobility group AT-Hook 2 (Hmga2) and Prom1. Moreover, OPCL61 formed a GBM-like brain tumor with features such as necrosis, hemorrhaging, and angiogenesis, even when just ten cells were injected into the brains of nude mice. These results indicate that pre-CPC, such as p53KO OPC, serve as a seed for CSC. Interestingly, it has been observed that p53 knockdown fibroblasts readily acquire pluripotency when Yamanaka factors are overexpressed [54–56], suggesting a significant connection between stemness and transformation.
Hide et al. isolated tumorigenic single CSC clones from H-RasL61-overexpressing p53KO NSC (NSCL61). These clones, when transplanted into the brains of nude mice, formed GBM-like brain tumors. These single CSCs were capable of generating both CSCs and differentiation marker-positive cells expressing either the neuronal marker microtubule-associated protein or the AST marker glial fibrillary acidic protein, both in culture and in vivo. This finding suggests that each CSC has the potential to generate heterogenous cancer cells within the tumor [57]. Additionally, the same group discovered that NSCL61 and OPCL61 exhibited different drug resistance capacities. While the combination of gefitinib and celecoxib inhibited tumorigenesis of OPCL61, it had no effect on NSCL61 [53]. This highlights that the difference of cell-of-origin of CSC is one of the factors contributing CSC heterogeneity and multiple drug resistance.
CSC heterogeneity has also been observed in mouse models, such as p53KO mice, which initially develop but eventually suffer from various types of cancer. Zhang et al. showed that breast cancer, which formed in p53KO mice, consisted of four different tumorigenic populations, characterized as Lineage–(Lin–)CD29high(CD29H)CD24H, Lin–CD29HCD24Low(CD24L), Lin–CD29LCD24H, and Lin–CD29LCD24L. This suggests that pre-CSC and/or pre-CPC transform into different types of CSCs over time [20]. Due to the presence of multiple CSC types with distinct mutations, these cancers are likely resistant to various therapies.
Additionally, single-cell RNA sequencing (scRNAseq) analysis has revealed the heterogeneity of GSCs in GBM specimen (see Table 2). Patel et al. demonstrated that individual GBMs contain four heterogenous GSCs: proneural, neural, classical and MES types, identifying using CD133 as a GSC marker. They also observed hybrid states with two subtypes, either classical and proneural or MES and neural, revealing the existence of GSCs with abnormal developmental programs [58].
Bhaduri et al. also demonstrated that single GBM contains multiple subtypes of GSCs, using four GSC markers: PROM1, Fucosyltransferase 4 (FUT4), CD44, L1 cell adhesion molecule (L1CAM) and SOX2. Among the subtypes, they focused on the outer radical glia-like GSCs (oR-GSCs), as radial glia has not been identified in adult brain even using scRNAseq analysis [59]. They demonstrated that the oR-GSCs contribute to the cellular composition and invasive behavior of glioblastoma.
Furthermore, Dirkse et al. found that GBM contains sixteen types of GSCs, which express different sets of GSC markers, CD133, CD44, A2B5 and CD15, in patient specimen. They demonstrated that hypoxia endows the cancer cells with the CSC status and that the phenotype is reversible [40]. They concluded that understanding the mechanism of phenotype transition is crucial for therapy.
Considering the heterogeneity of CSC, including the diversity of cells-of-origin, reversion to CSC, cell-cell interaction, environment stress, and varying therapy resistance, characterizing the heterogeneity in whole cancers will lead to the development of new therapeutic strategies.
4. Potential therapeutic methods for overcoming CSC heterogeneity
Given the heterogeneity of cancer, the question is how we can develop novel therapeutic methods to conquer it. One promising approach is the use of cancer organoids directly prepared from patient specimens. These organoids likely consist of CSCs, cancer cells, and surrounding non-cancer cells [60,61]. Nonetheless, cancer organoids do not completely mirror original cancers because tumor-surrounding non-cancerous cells, such as immune cells, and systemic factors in the bloodstream are not continuously present in culture. There is no data regarding which culture conditions can maintain whole cancer cell heterogeneity in vitro. Based on the available methods, we cannot exclude the possibility that the defined culture medium selects specific cells, such as adaptable CSCs and their sister cells, for survival.
Although scRNAseq reveals cancer heterogeneity using directly prepared patient specimens, these data do not provide the essential information on which cells retain tumorigenicity and are potential therapeutic targets, because CSC marker-negative cells, such as CD133-negative cancer cells, have also been shown to be tumorigenic when transplanted into immunodeficient mice [62]. Thus, without establishing methods that can determine the phenotype of each single cell in a tumor, it is difficult to generate a useful CSC heterogeneity profile that may be used to develop tailor-made therapeutic strategies targeting each CSC type. Therefore, targeting common CSC factors is likely to be a realistic therapeutic method at this time if the off-target effects on normal cells, particular normal tissue-specific stem cells, can be minimized.
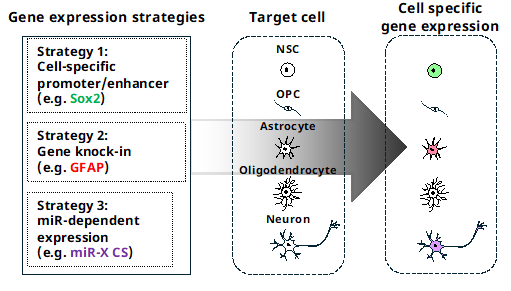
Gene therapy is widely used to treat various disorders. It can be used to manipulate target gene (knockdown or KO), overexpress exogenous genes in target cells using specific promoters and/or enhancers, and deliver therapeutic genes using viruses [63]. Compared to antibody-based therapeutic methods or the identification of chemicals to target disease specifically, constructing a vector for gene therapy is much easier. However, if there is no specific promoter or enhancer to target cells specifically, it is impossible to express exogenous genes only in target cells. Additionally, the large size of the promoter and enhancer or having multiple promoters/enhancers poses another obstacle to successful gene therapy targeting specific cells.
Naldini and his colleagues established a new method that specifically regulates the expression of an exogenous gene only in the target cells in a microRNA (miR) expression-dependent manner (Figure 3). They elegantly demonstrated that endogenously expressed miRs, miR-142-3p specific to hematopoietic cells and miR-302a specific to human embryonic stem cells, suppress the expression of an exogenous gene when combined with four tandem complementary sequences of the miRs (miRCS) in vitro and in vivo [64,65].
Sayeg et al. also showed that miRCS of miR-128 and miR-221, both expressed in excitatory neurons, but not in inhibitory neurons, selectively induced the expression of an exogenous gene in inhibitory neurons in the brain when the expression vector was delivered with miRCS using a lentivirus [66].
Using miR-regulated expression system, Suzuki et al. have demonstrated that the herpes simplex virus encoding the thymidine kinase with target sequences of miR-122a, which expresses in hepatocytes, killed the tumor in liver with little hepatotoxicity, when injected intratumorally [67].
These findings suggest that posttranscriptional gene silencing by endogenous miR can specifically induce the expression of any exogenous gene with miRCS in miR-negative target cells, but not miR-positive others.
The miRNA Tissue Atlas (https://ccb-web.cs.uni-saarland.de/tissueatlas2/) revealed that the expression of miR-142-3p is highest in the lung, followed by the brain, whereas the expression of both miR-128 and miR-221 is low in non-brain tissues. This suggests that miRCS alone may not be enough regulate the expression of an exogenous gene restricted to the target cells in vivo. Nonetheless, miR-dependent gene expression systems are very attractive for generating new therapeutics by combining them with other innovative ideas and technologies.
A remaining question is how to selectively deliver the expression system selectively into CSCs. Various methods have been developed for delivering therapeutics into tumors [68]. In the case of brain tumors, different delivery methods such as viral vectors, liposomes, cationic polymers, gene guns, and cell-penetrating peptides have been evaluated [69]. Among these methods, dual-targeting nanoparticles that can pass through the blood–brain barrier (BBB) and target brain tumors show promise. However, there is a concern that these particles may also deliver chemicals to other tissues, particularly the liver [70].
Another potential option is the use of recombinant viruses, including Adeno-Associated Virus (AAV), Zika virus, and Herpes simplex virus, which can infect brain cancer cells and activate a cytotoxic signal to kill them in vivo [71–74]. AAV seems to be superior due to its low immune response, high yield, long-term expression in nonproliferating cells, and histotrophic ability [75,76]. Recent studies have identified brain tropic AAV capsids, such as PHP.eB, CAP-B10, and CAP-B22, which can target the brain and enable the analysis of gene function in both endogenous and exogenous gene in mice and nonhuman primates. However, it was found that these capsids also deliver a small number of virus particles to the liver [77,78]. Although AAV is not effective for long-term expression in rapidly dividing cells, such as cancer cells, due to its low genome integration, this problem could potentially be addressed by using a Cas9-dependent genome-editing (GE) system, as its transient expression is sufficient to knockout the target gene [79].
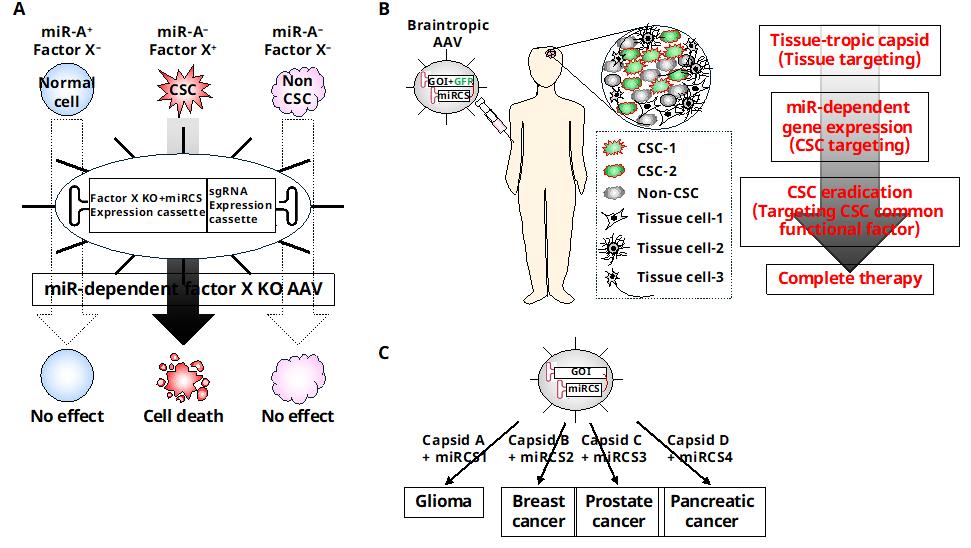
By combining key factors of CSC function, selective miR-dependent gene expression systems specific to CSC, and AAV-based tissue targeting, it may be possible to eliminate different types of heterogeneous CSCs in tumors without causing harm to normal cells or non-CSCs (Figure 4). Recent research by Wan et al. has shown promising results in this area. They developed brain tropic AAVs that encode a miR-dependent GE system specifically targeting a common functional factor in GSC. Their experiments demonstrated that the virus selectively transduced the system into GSCs in the brain without affecting other tissues, including the liver, and effectively killed the GSCs in vivo when administered intravenously (unpublished observation). Although the tumor was not completely cured in the treated mice, further improvements to this system are needed before it can be translated into the clinical setting. Nevertheless, this approach shows promise not only for eliminating different types of GSCs and overcoming CSC heterogeneity but also for eradicating various types of cancers by replacing the AAV capsid, miRCS, and CSC target genes.
5. Conclusions and perspectives
Technological advances, such as scRNAseq, CyTOF and special transcriptomics analysis, have revealed the heterogeneity of tumors, which consist of various types of cancer cells and non-cancer cells. However, this information alone is insufficient to develop therapeutic methods that target heterogeneous CSC, as there is currently no way to verify which cells retain tumorigenicity in vivo. As a result, a more realistic therapeutic approach is to target common CSC factors while minimizing the impact on normal cells, particularly ts-SC/PCs. One potential method is to use a tissue-tropic capsid-coated AAV that encodes a miR-dependent GE system for CSC factors. This AAV can be delivered to the target tissue and the GE system will only be expressed only in CSCs in a miR-dependent manner, effectively knocking out key CSC functional factor and eliminating heterogeneous CSCs without significant side effects.
Declarations
Ethics Statement
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Not applicable.
Funding
T.K. was supported in part by an AMED Practical Research for Innovative Cancer Control (17ck0106236h0002) and a Grant-In-Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan (20H03559).
Competing Interests
The author has declared that no competing interests exist.
| Typical CSC marker |
Cancer type |
Enrichment method |
Function |
References |
| CD133 (Prominin 1, Prom1) |
Multiple cancer, including Glioblastoma, Colorectal cancer |
Flow cytometry |
Survival and proliferation |
[12-17] |
| CD44 |
Multiple cancer, including Breast cancer, Head and neck cancer, Glioblastoma |
Flow cytometry |
Cell-cell interaction, Activation of PI3K-4EBP1-SOX2 pathway by binding with hyaluronic acid |
[18,19] |
| CD24 |
Multiple cancer, including Breast cancer, Colorectal cancer |
Flow cytometry |
B-cell activation and proliferation, ERK1/2activation |
[20,21] |
| CD49f (Integrin subunit alpha 6, ITGA6) |
Multiple cancer, including Breast cancer, Glioblastoma |
Flow cytometry |
Multiple functions, including laminin receptor, sperm-egg fusion and Insulin growth factor signalling |
[21–23] |
| CD15 (Lewis x, Fucosyltransferase4 (FUT4), Stage-Specific Embryonic Antigen 1 (SSEA1) |
Glioblastoma, Medulloblastoma |
Flow cytometry |
Cell-cell interaction, adhesion |
[24,25] |
| LGR5 (Leucine Rich Repeat Containing G Protein-Coupled Receptor 5, G-Protein Coupled Receptor 49 (GPR49) |
Multiple cancer, including Colorectal cancer, Gastric cancer, Liver cancer |
Flow cytometry |
Wnt signal |
[26,27] |
| ABC transporters (e.g. ABCG2) |
Multiple cancer, including Acute myeloid leukemia, Breast cancer, Glioblastoma, Colorectal cancer, Pancreatic cancer, etc. |
Flow cytometry |
Exclusion of chemicals and dyes, such asHoechst33342 and cytotoxic drugs |
[28,29] |
| Aldehyde Dehydrogenase (ALDH) |
Multiple cancer, including Acute myeloid leukemia, Breast cancer, Glioblastoma, Colorectal cancer, Pancreatic cancer, etc. |
Flow cytometry |
Detoxification |
[30,31] |
Table 2 GSC heterogeneity.
| Number of heterogeneous GSC in cancer |
GSC markers |
GBM microenvironment |
Major finding |
Reference |
| Four (proneural, neural, classical mesenchymal) |
CD133 (PROM1) |
Hypoxia |
Different types of GSCs co-existed in one GBM. |
[58] |
| Two to seven |
SOX2, FUT4, PROM1, CD44 L1CAM |
|
Outer radial glial-like cancer cell was identified as a new GSC. |
[59] |
| Sixteen |
CD133, CD44, A2B5, CD15 |
Hypoxia |
Heterogeneity arised from reversible state transitions instructed by the microenvironment. |
[40] |
References
| 1. |
Weinberg RA. The Biology of Cancer. New York: W.W. Norton & Company; 2003.
[CrossRef]
|
| 2. |
Lawson DA, Kessenbrock K, Davis RT, Pervolarakis N, Werb Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat Cell Biol. 2018;20(12):1349-1360.
[Google Scholar]
[CrossRef]
|
| 3. |
Abdelfattah N, Kumar P, Wang C, Leu JS, Flynn WF, Gao R, et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat Commun. 2022;13 Supplement_1:767.
[Google Scholar]
[CrossRef]
|
| 4. |
Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. 2017;16:41.
[Google Scholar]
[CrossRef]
|
| 5. |
Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. 2011;108(19):7950-7955.
[Google Scholar]
[CrossRef]
|
| 6. |
Mani S A, Guo W, Liao M J, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704-715.
[Google Scholar]
[CrossRef]
|
| 7. |
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645-648.
[Google Scholar]
[CrossRef]
|
| 8. |
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-111.
[Google Scholar]
[CrossRef]
|
| 9. |
Singh SK, Clarke ID, Hide T, Dirks PB. Cancer stem cells in nervous system tumors. Oncogene. 2004;23(43):7267-7273.
[Google Scholar]
[CrossRef]
|
| 10. |
Kondo T. Brain cancer stem-like cells. Eur J Cancer. 2006;42:1237-1242.
[Google Scholar]
[CrossRef]
|
| 11. |
Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6:225-236.
[Google Scholar]
[CrossRef]
|
| 12. |
Corbeil D, Roper K, Hellwig A, Tavian M, Miraglia S, Watt SM, et al. The human AC133 hematopoietic stem cell antigen is also expressed in epithelial cells and targeted to plasma membrane protrusions. J Biol Chem. 2000;275(8):5512-5520.
[Google Scholar]
[CrossRef]
|
| 13. |
Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, et al. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci USA. 2000;97:14720-14725.
[Google Scholar]
[CrossRef]
|
| 14. |
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821-5828.
[Google Scholar]
|
| 15. |
Zelhof AC, Hardy RW, Becker A, Zuker CS. Transforming the architecture of compound eyes. Nature. 2006;443(7112):696-699.
[Google Scholar]
[CrossRef]
|
| 16. |
Tárnok A, Ulrich H, Bocsi J. Phenotypes of stem cells from diverse origin. Cytometry A. 2001;77A:6-10.
[Google Scholar]
[CrossRef]
|
| 17. |
Dellett M, Sasai N, Nishide K, Becher S, Papadaki V, Limb GA, et al. Genetic background and light-dependent progression of photoreceptor cell degeneration in prominin-1 knockout mice. Invest. Ophthalmol. Vis Sci. 2014;56:164-176.
[Google Scholar]
[CrossRef]
|
| 18. |
Naor D, Sionov RV, Ish-Shalom D. CD44: structure, function, and association with the malignant process. Adv Cancer Res. 1997;71:241-319.
[Google Scholar]
[CrossRef]
|
| 19. |
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983-3988.
[Google Scholar]
[CrossRef]
|
| 20. |
Zhang M, Behbod F, Atkinson RL, Landis MD, Kittrell F, Edwards D, et al. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 2008;68(12):4674-4682.
[Google Scholar]
[CrossRef]
|
| 21. |
Vassilopoulos A, Chisholm C, Lahusen T, Zheng H, Deng CX. A critical role of CD29 and CD49f in mediating metastasis for cancer-initiating cells isolated from a Brca1-associated mouse model of breast cancer. Oncogene. 2013;33(47):5477-5482.
[Google Scholar]
[CrossRef]
|
| 22. |
Kumagai Y, Naoki H, Nakasyo E, Kamioka Y, Kiyokawa E, Matsuda M. Heterogeneity in ERK activity as visualized by in vivo FRET imaging of mammary tumor cells developed in MMTV-Neu mice. Oncogene. 2014;34(8):1051-1057.
[Google Scholar]
[CrossRef]
|
| 23. |
Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. 2010;6(5):421-432.
[Google Scholar]
[CrossRef]
|
| 24. |
Read TA, Fogarty MP, Markant SL, McLendon RE, Wei Z, Ellison DW, et al. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell. 2009;15(2):135-147.
[Google Scholar]
[CrossRef]
|
| 25. |
Son MJ, Woolard K, Nam DH, Lee J, Fine HA. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. 2009;4(5):440-452.
[Google Scholar]
[CrossRef]
|
| 26. |
Merlos-Suárez A, Barriga FM, Jung P, Iglesias M, Céspedes MV, Rossell D, et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. 2011;8(5):511-524.
[Google Scholar]
[CrossRef]
|
| 27. |
Li XB, Yang G, Zhu L, Tang YL, Zhang C, Ju Z, et al. Gastric Lgr5(+) stem cells are the cellular origin of invasive intestinal-type gastric cancer in mice. Cell Res. 2016;26(7):838-849.
[Google Scholar]
[CrossRef]
|
| 28. |
Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci U S A. 2004;101(3):781-786.
[Google Scholar]
[CrossRef]
|
| 29. |
Leccia F, Del Vecchio L, Mariotti E, Di Noto R, Morel AP, Puisieux A, et al. ABCG2, a novel antigen to sort luminal progenitors of BRCA1- breast cancer cells. Mol Cancer. 2014;13:213.
[Google Scholar]
[CrossRef]
|
| 30. |
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555-567.
[Google Scholar]
[CrossRef]
|
| 31. |
Yeo SK, Wen J, Chen S, Guan JL. Autophagy Differentially Regulates Distinct Breast Cancer Stem-like Cells in Murine Models via EGFR/Stat3 and Tgfβ/Smad Signaling. Cancer Res. 2016;76(11):3397-3410.
[Google Scholar]
[CrossRef]
|
| 32. |
White RM, Cech J, Ratanasirintrawoot S, Lin CY, Rahl PB, Burke CJ, et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471(7339):518-522.
[Google Scholar]
[CrossRef]
|
| 33. |
Sykes DB, Kfoury YS, Mercier FE, Wawer MJ, Law JM, Haynes MK, et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell. 2016;167(1):171-186.e15.
[Google Scholar]
[CrossRef]
|
| 34. |
Brown KK, Spinelli JB, Asara JM, Toker A. Adaptive reprogramming of de novo pyrimidine synthesis is a metabolic vulnerability in triple-negative breast cancer. Cancer Discov. 2017;7(4):391-399.
[Google Scholar]
[CrossRef]
|
| 35. |
Koundinya M, Sudhalter J, Courjaud A, Lionne B, Touyer G, Bonnet L, et al. Dependence on the Pyrimidine Biosynthetic Enzyme DHODH Is a Synthetic Lethal Vulnerability in Mutant KRAS-Driven Cancers. Cell Chem Biol. 2018;25(6):705-717.
[Google Scholar]
[CrossRef]
|
| 36. |
Ladds M, van Leeuwen IMM, Drummond CJ, Chu S, Healy AR, Popova G, et al. A DHODH inhibitor increases p53 synthesis and enhances tumor cell killing by p53 degradation blockage. Nat Commun. 2018;9:2071.
[Google Scholar]
[CrossRef]
|
| 37. |
Wang X, Yang K, Wu Q, Kim LJY, Morton AR, Gimple RC, et al. Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells. Sci Transl Med. 2019;11(504):eaau4972.
[Google Scholar]
[CrossRef]
|
| 38. |
Echizenya S, Ishii Y, Kitazawa S, Tanaka T, Matsuda S, Watanabe E, et al. Discovery of a new pyrimidine synthesis inhibitor eradicating glioblastoma-initiating cells. Neuro Oncol. 2020;22(2):229-239.
[Google Scholar]
[CrossRef]
|
| 39. |
Breedveld FC, Dayer JM. Leflunomide: mode of action in the treatment of rheumatoid arthritis. Ann Rheum Dis. 2000;59(11):841-849.
[Google Scholar]
[CrossRef]
|
| 40. |
Dirkse A, Golebiewska A, Buder T, Nazarov PV, Muller A, Poovathingal S, et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019;10:1787.
[Google Scholar]
[CrossRef]
|
| 41. |
Kondo T, Raff M. Oligodendrocyte precursor cells reprogrammed to become multipotential CNS stem cells. Science. 2000;289(5485):1754-1757.
[Google Scholar]
[CrossRef]
|
| 42. |
Laywell ED, Rakic P, Kukekov VG, Holland EC, Steindler DA. Identification of a multipotent astrocytic stem cell in the immature and adult mouse brain. Proc Natl Acad Sci U S A. 2000;97(25):13883-13888.
[Google Scholar]
[CrossRef]
|
| 43. |
Hu P, Zhang W, Xin H, Deng G. Single Cell Isolation and Analysis. Front Cell Dev Biol. 2016;4:116.
[Google Scholar]
[CrossRef]
|
| 44. |
Mohan A, Raj Rajan R, Mohan G, Kollenchery Puthenveettil P, Maliekal TT. Markers and Reporters to Reveal the Hierarchy in Heterogenous Cancer Stem Cells. Front Cell Dev Biol. 2021;9:668851.
[Google Scholar]
[CrossRef]
|
| 45. |
Vegliante R, Pastushenko I, Blanpain C. Deciphering functional tumor states at single-cell resolution. EMBO J. 2022;41(2):e109221.
[Google Scholar]
[CrossRef]
|
| 46. |
Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019;178(4):835-849.
[Google Scholar]
[CrossRef]
|
| 47. |
Abdelfattah N, Kumar P, Wang C, Leu JS, Flynn WF, Gao R, et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat Commun. 2022;13:767.
[Google Scholar]
[CrossRef]
|
| 48. |
van den Bent MJ, Geurts M, French PJ, Smits M, Capper D, Bromberg JEC, et al. Primary brain tumours in adults. Lancet. 2023;402(10412):1564-1579.
[Google Scholar]
[CrossRef]
|
| 49. |
Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, Dykgers A, et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell. 2019;177(5):1330-1345.
[Google Scholar]
[CrossRef]
|
| 50. |
Yu K, Hu Y, Wu F, Guo Q, Qian Z, Hu W, et al. Surveying brain tumor heterogeneity by single-cell RNA-sequencing of multi-sector biopsies. Natl Sci Rev. 2020;7:1306-1318.
[Google Scholar]
[CrossRef]
|
| 51. |
Ravi VM, Will P, Kueckelhaus J, Sun N, Joseph K, Salié H, et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell. 2022;40:639-655.
[Google Scholar]
[CrossRef]
|
| 52. |
Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17(4):362-375.
[Google Scholar]
[CrossRef]
|
| 53. |
Hide T, Takezaki T, Nakatani Y, Nakamura H, Kuratsu J, Kondo T. Combination of a ptgs2 inhibitor and an epidermal growth factor receptor-signaling inhibitor prevents tumorigenesis of oligodendrocyte lineage-derived glioma-initiating cells. Stem Cells. 2011;29(4):590-599.
[Google Scholar]
[CrossRef]
|
| 54. |
Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460(7259):1132-1135.
[Google Scholar]
[CrossRef]
|
| 55. |
Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460(7259):1140-1144.
[Google Scholar]
[CrossRef]
|
| 56. |
Li H, Collado M, Villasante A, Strati K, Ortega S, Cañamero M, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460(7259):1136-1139.
[Google Scholar]
[CrossRef]
|
| 57. |
Hide T, Takezaki T, Nakatani Y, Nakamura H, Kuratsu J, Kondo T. Sox11 prevents tumorigenesis of glioma-initiating cells by inducing neuronal differentiation. Cancer Res. 2009;69(20):7953-7959.
[Google Scholar]
[CrossRef]
|
| 58. |
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396-1401.
[Google Scholar]
[CrossRef]
|
| 59. |
Bhaduri A, Di Lullo E, Jung D, Müller S, Crouch EE, Espinosa CS, et al. Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. 2020;26(1):48-63.e6.
[CrossRef]
|
| 60. |
Kondo J, Endo H, Okuyama H, Ishikawa O, Iishi H, Tsujii M, et al. Retaining cell-cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. Proc Natl Acad Sci USA. 2011;108(15):6235-6240.
[Google Scholar]
[CrossRef]
|
| 61. |
Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340(6137):1190-1194.
[Google Scholar]
[CrossRef]
|
| 62. |
Vermeulen L, Todaro M, de Sousa Mello F, Sprick MR, Kemper K, Perez Alea M, et al. Single-cell cloning of colon cancer stem cells reveals a multi-linega differentiation capacity. Proc Natl Acad Sci USA. 2008;105(36):13427-13432.
[Google Scholar]
[CrossRef]
|
| 63. |
High KA, Roncarolo MG. Gene Therapy. N Engl J Med. 2019;381(5):455-464.
[Google Scholar]
[CrossRef]
|
| 64. |
Brown BD, Venneri MA, Zingale A, Sergi L, Naldini L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat Med. 2006;12(5):585-591.
[Google Scholar]
[CrossRef]
|
| 65. |
Brown BD, Gentner B, Cantore A, Colleoni S, Amendola M, Zingale A, et al. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat Biotechnol. 2007;25(12):1457-1467.
[Google Scholar]
[CrossRef]
|
| 66. |
Sayeg MK, Weinberg BH, Cha SS, Goodloe M, Wong WW, Han X. Rationally designed microRNA-based genetic classifiers target specific neurons in the brain. ACS Synth Biol. 2015;4(7):788-795.
[Google Scholar]
[CrossRef]
|
| 67. |
Suzuki T, Sakurai F, Nakamura S, Kouyama E, Kawabata K, Kondoh M, et al. miR-122a-regulated expression of a suicide gene prevents hepatotoxicity without altering antitumor effects in suicide gene therapy. Mol Ther. 2008;16:1719-1726.
[Google Scholar]
[CrossRef]
|
| 68. |
Butt MH, Zaman M, Ahmad A, Khan R, Mallhi TH, Hasan MM, et al. Appraisal for the potential of viral and nonviral vectors in gene therapy: A review. Genes (Basel). 2022;13(8):1370.
[Google Scholar]
[CrossRef]
|
| 69. |
Goswami R, Subramanian G, Silayeva L, Newkirk I, Doctor D, Chawla K, et al. Gene therapy leaves a vicious cycle. Front Oncol. 2019;9:297.
[Google Scholar]
[CrossRef]
|
| 70. |
Farshbaf M, Mojarad-Jabali S, Hemmati S, Khosroushahi AY, Motasadizadeh H, Zarebkohan A, et al. Enhanced BBB and BBTB penetration and improved anti-glioma behavior of Bortezomib through dual-targeting nanostructured lipid carriers. J Control Release. 2022;345:371-384.
[Google Scholar]
[CrossRef]
|
| 71. |
Todo T, Rabkin SD, Sundaresan P, Wu A, Meehan KR, Herscowitz HB, et al. Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication-competent herpes simplex virus. Hum Gene Ther. 1999;10(17):2741-2755.
[Google Scholar]
[CrossRef]
|
| 72. |
Zhu Z, Gorman MJ, McKenzie LD, Chai JN, Hubert CG, Prager BC, et al. Zika virus has oncolytic activity against glioblastoma stem cells. J Exp Med. 2017;214(10):2843-2857.
[Google Scholar]
[CrossRef]
|
| 73. |
Kardani K, Gil JS, Rabkin SD. Oncolytic herpes simplex viruses for the treatment of glioma and targeting glioblastoma stem-like cells. Front Cell Infect Microbiol. 2023;13:1206111.
[Google Scholar]
[CrossRef]
|
| 74. |
Calderón-Peláez MA, Maradei Anaya SJ, Bedoya-Rodríguez IJ, González-Ipuz KG, Vera-Palacios D, Buitrago IV, et al. Zika Virus: A Neurotropic Warrior against High-Grade Gliomas-Unveiling Its Potential for Oncolytic Virotherapy. Viruses. 2024;16(4):561.
[Google Scholar]
[CrossRef]
|
| 75. |
Pupo A, Fernández A, Low SH, François A, Suárez-Amarán L, Samulski RJ. AAV vectors: The Rubik’s cube of human gene therapy. Mol Ther. 2022;30(12):3515-3541.
[Google Scholar]
[CrossRef]
|
| 76. |
Ling Q, Herstine JA, Bradbury A, Gray SJ. AAV-based in vivo gene therapy for neurological disorders. Nat Rev Drug Discov. 2023;22(10):789-806.
[Google Scholar]
[CrossRef]
|
| 77. |
Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. 2017;20(8):1172-1179.
[Google Scholar]
[CrossRef]
|
| 78. |
Goertsen D, Flytzanis NC, Goeden N, Chuapoco MR, Cummins A, Chen Y, et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat Neurosci. 2022;25:106-115.
[Google Scholar]
[CrossRef]
|
| 79. |
Brommel CM, Cooney AL, Sinn PL. Adeno-Associated Virus-Based Gene Therapy for Lifelong Correction of Genetic Disease. Hum Gene Ther. 2020;31(17–18):985-995.
[Google Scholar]
[CrossRef]
|