
Human Population Genetics and Genomics ISSN 2770-5005
Human Population Genetics and Genomics 2026;6(2):0007 | https://doi.org/10.47248/hpgg2606020007
Original Research Open Access
The reliability of inferred archaic segments
Nancy Bird

 ,
Erin Walker
,
Garrett Hellenthal
,
Erin Walker
,
Garrett Hellenthal
Correspondence: Nancy Bird; Garrett Hellenthal
Academic Editor(s): Joshua Akey
Received: Aug 31, 2025 | Accepted: Apr 27, 2026 | Published: May 15, 2026
Cite this article: Bird N, Walker E, Hellenthal G. The reliability of inferred archaic segments. Hum Popul Genet Genom. 2026;6(2):0007. https://doi.org/10.47248/hpgg2606020007
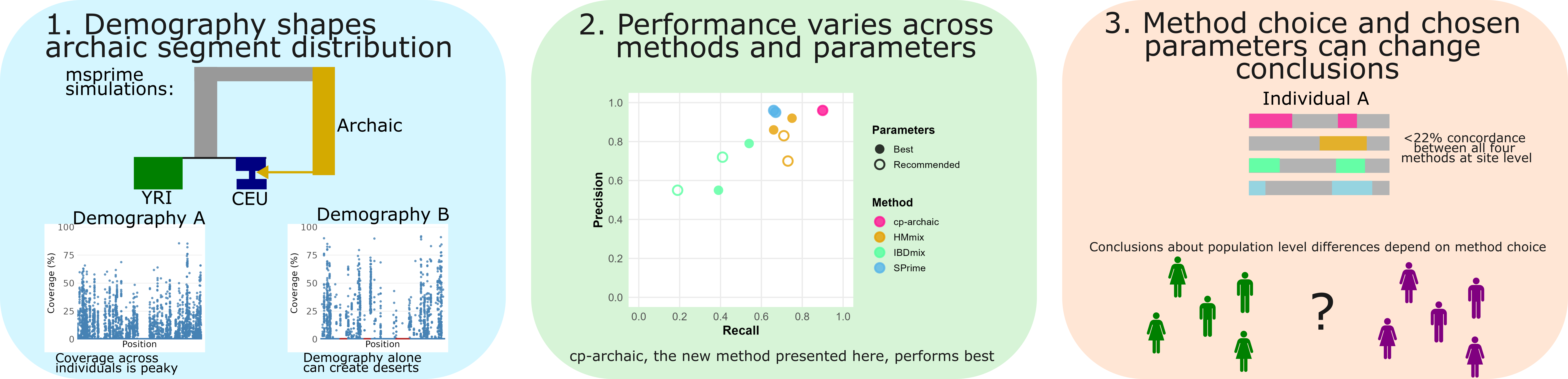
Many or all present-day human genomes carry segments of DNA inherited from archaic humans due to admixture events occurring >25,000 years ago. Several methods have been published to detect such segments. These variously require phased haplotypes, an archaic reference sequence and/or an outgroup with little related archaic introgression. Inference from these approaches have been used to document purifying and positive selection of archaic segments and to understand their influence on the phenotype. However, the comparative accuracy of different methods at detecting archaic segments, and how well inferred segments overlap across methods, remains underexplored. Here, we used demographic simulations to evaluate accuracy in detecting archaic ancestry under three widely-used approaches: SPrime, IBDMix and HMmix, and a technique introduced here, cp-archaic, as well as applying all approaches to 1000 Genomes data. Our results reveal substantial variation in method performance, with recall and precision ranges differing significantly across approaches and parameter settings. cp-archaic achieved the highest accuracy overall (F1, the harmonic mean of precision and recall=0.92-0.94 across three different simulations). The choice of demographic simulation substantially impacted the distribution and characteristics of true archaic segments, with demographic scenarios creating both archaic ancestry ‘deserts’ and regions of very high archaic ancestry in the absence of selection. Notably, we also found low agreement between methods, with <22% overlap in individual archaic sites detected across all four approaches in real data. These findings highlight how conclusions about archaic introgression patterns, population differences, and genome-wide coverage depend critically on both methodological choice and underlying demographic assumptions. We recommend careful method selection and parameter optimisation, as well as caution when interpreting individual archaic segments, particularly in comparative studies across populations.
Keywordsarchaic introgression, Neanderthal, methods, local ancestry inference
Share this article




![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy

