
Human Population Genetics and Genomics ISSN 2770-5005
Human Population Genetics and Genomics 2026;6(2):0004 | https://doi.org/10.47248/hpgg2606020004
Technical Note Open Access
Clumppling 2.0: A Clustering Alignment Program for Population Structure Analyses
Xiran Liu
1

 ,
Noah A. Rosenberg
2
,
Sohini Ramachandran
1,3
,
Noah A. Rosenberg
2
,
Sohini Ramachandran
1,3
Correspondence: Xiran Liu
Academic Editor(s): Joshua Akey, Carina Schlebusch, Torsten Günther
Received: Nov 21, 2025 | Accepted: Feb 24, 2026 | Published: Mar 17, 2026
© 2026 by the author(s). This is an Open Access article distributed under the Creative Commons License Attribution 4.0 International (CC BY 4.0) license, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: Liu X, Rosenberg NA, Ramachandran S. Clumppling 2.0: A Clustering Alignment Program for Population Structure Analyses. Hum Popul Genet Genom. 2026;6(2):0004. https://doi.org/10.47248/hpgg2606020004
We previously introduced
admixture, alignment, clustering, genetic ancestry, population structure
Population structure analysis uses clustering methods on genotype data to model individual genomes as mixtures of contributions from multiple sources or latent clusters. When prior information about cluster membership is unavailable, these clusters are inferred directly from genotype data using unsupervised mixed-membership clustering [1–3]. Methods for this inference include
Label-switching. An unsupervised clustering method is equally likely to produce any of the K! permutations of the same set of cluster labels. These solutions are identical after appropriate permutation.
Multi-modality. Clustering runs can also produce truly distinct solutions (i.e., “modes”) that are not equivalent under any label permutation (e.g., Figure 1A). Multimodality can arise from the convergence of the clustering algorithm to different local or global optima, all providing plausible explanations for the data.

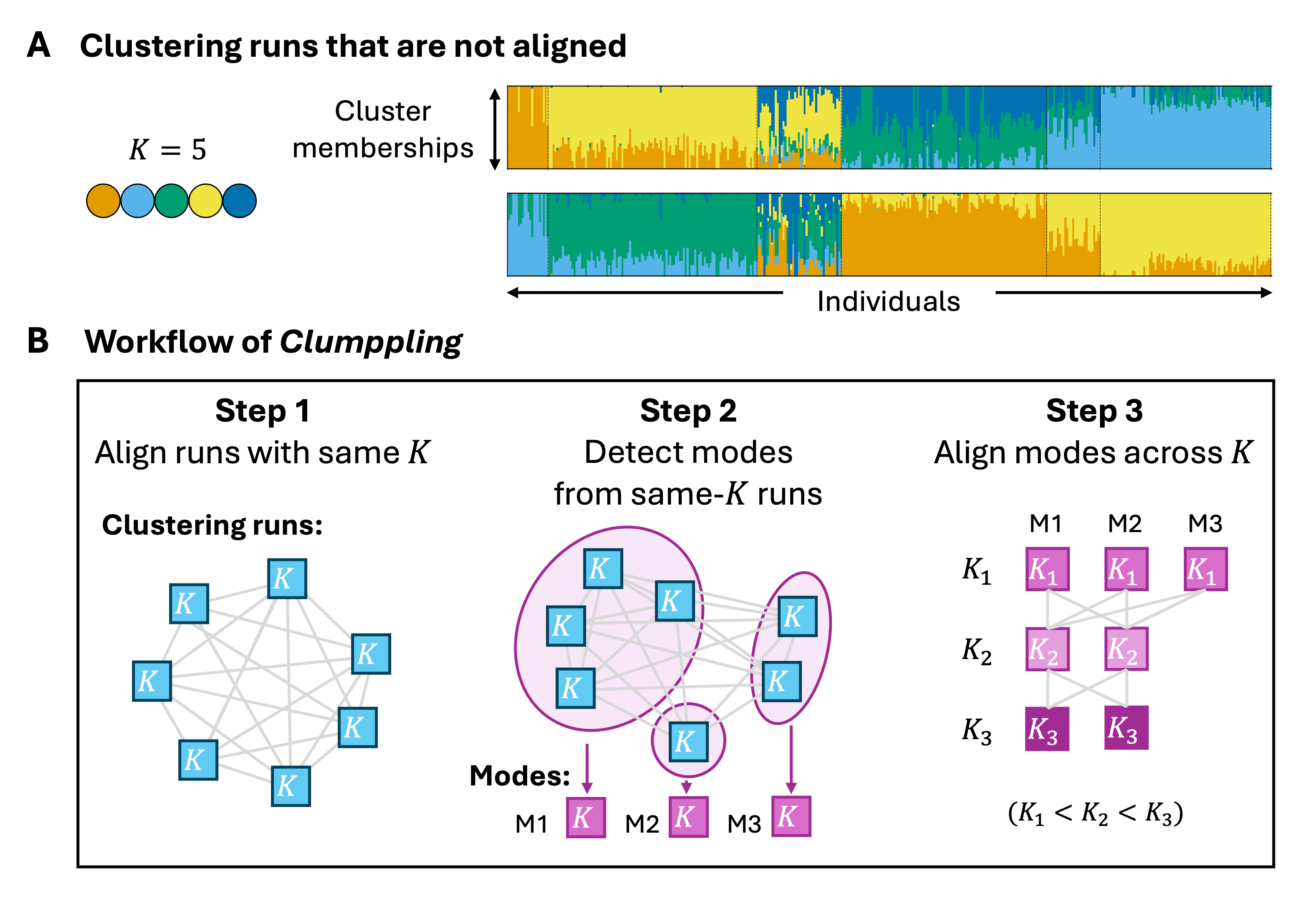
Figure 1. Unaligned clustering results and the workflow of Clumppling. (A) Bar plots from two example runs with K = 5 clusters, illustrating substantially different results. Unaligned plots are difficult to compare. (B) The workflow of
Different numbers of clusters (K) across runs. In unsupervised clustering, the number of clusters (K) typically needs to be specified by the user or inferred by the algorithm. Researchers often explore a range of values for K, but selecting a single optimal value is challenging and risks overlooking aspects of the population structure that are salient only at other values of K. Quantitatively examining results of multiple K values simultaneously can also be challenging, motivating alignment of runs across different K.
Multiple approaches have been developed to address these clustering alignment challenges, including
First,
Next,
Finally,
The results of a single clustering run are often visualized as a bar plot, where each individual’s membership coefficients across the K clusters are displayed as stacked, color-coded bars (see Figure 1A for examples with K = 5). To display its aligned clustering results,
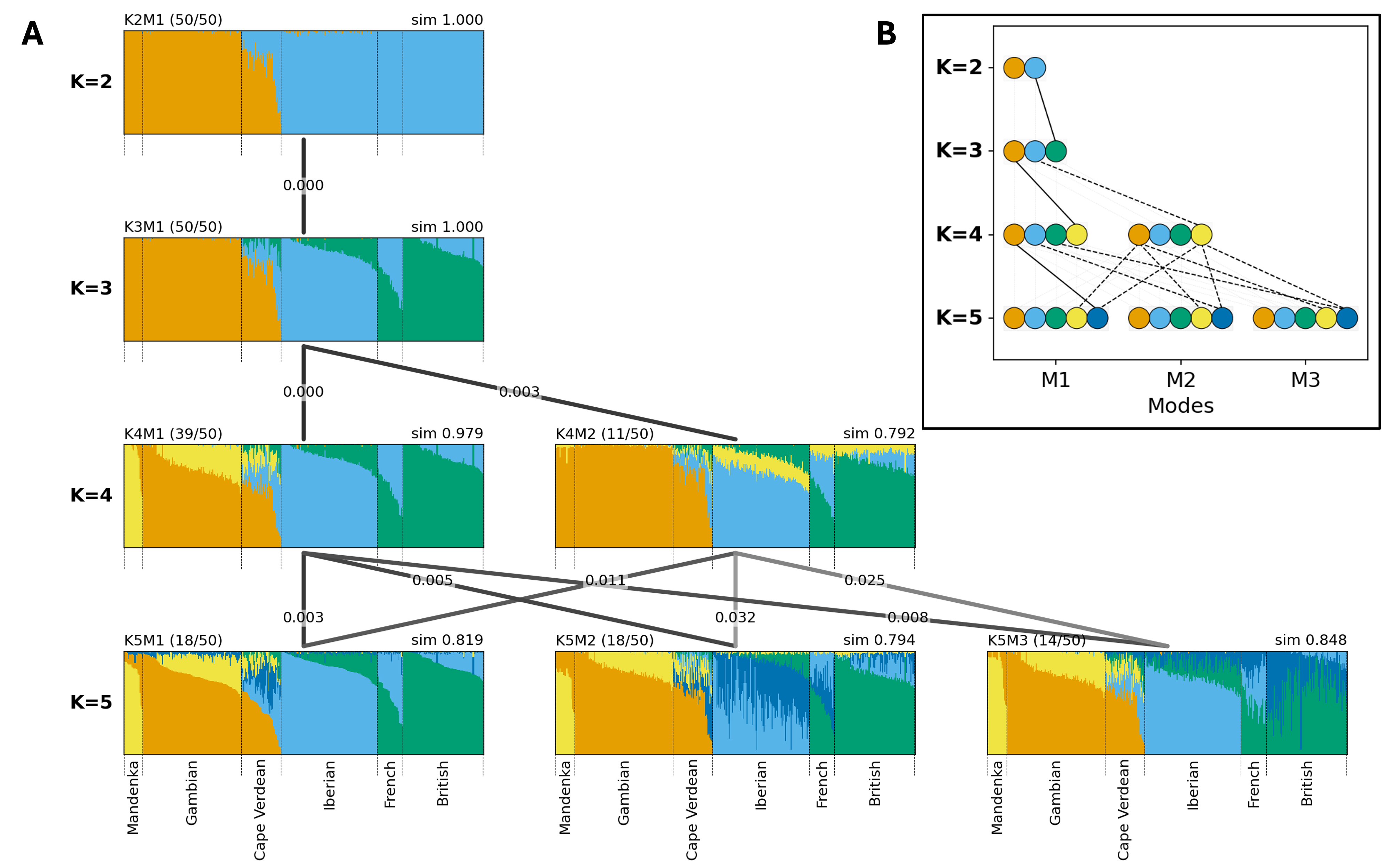
Figure 2. Clustering results from Cape Verde data aligned by Clumppling. There are 50 clustering runs for each K from 2 to 5. Detected modes are labeled, for example, “K2M1” (mode 1 of K = 2). (A) Clumppling’s multipartite graph of bar plots, showing the aligned memberships in each mode. For each bar plot, the mode size (i.e., the number of clustering runs contained in that mode) of all runs with the given K is shown in parentheses at the upper left, and the within-mode alignment similarity is shown at the upper right (e.g., sim=0.979). Modes with numbers of clusters K and K + 1 are connected by lines, where edge labels indicate alignment cost and edge color reflects alignment similarity. Darker edges indicate mode pairs with higher similarity, whereas lighter edges indicate poorer alignment. Individuals are grouped and labeled according to their population (provided as auxiliary labels by the user). Within each label group, individuals are displayed in decreasing order (left to right) by their membership in the cluster with the largest total membership in one specific plot (here K5M1); individuals are placed in this same order for all modes and all K. (B) The alignment pattern graph in
Another new feature in
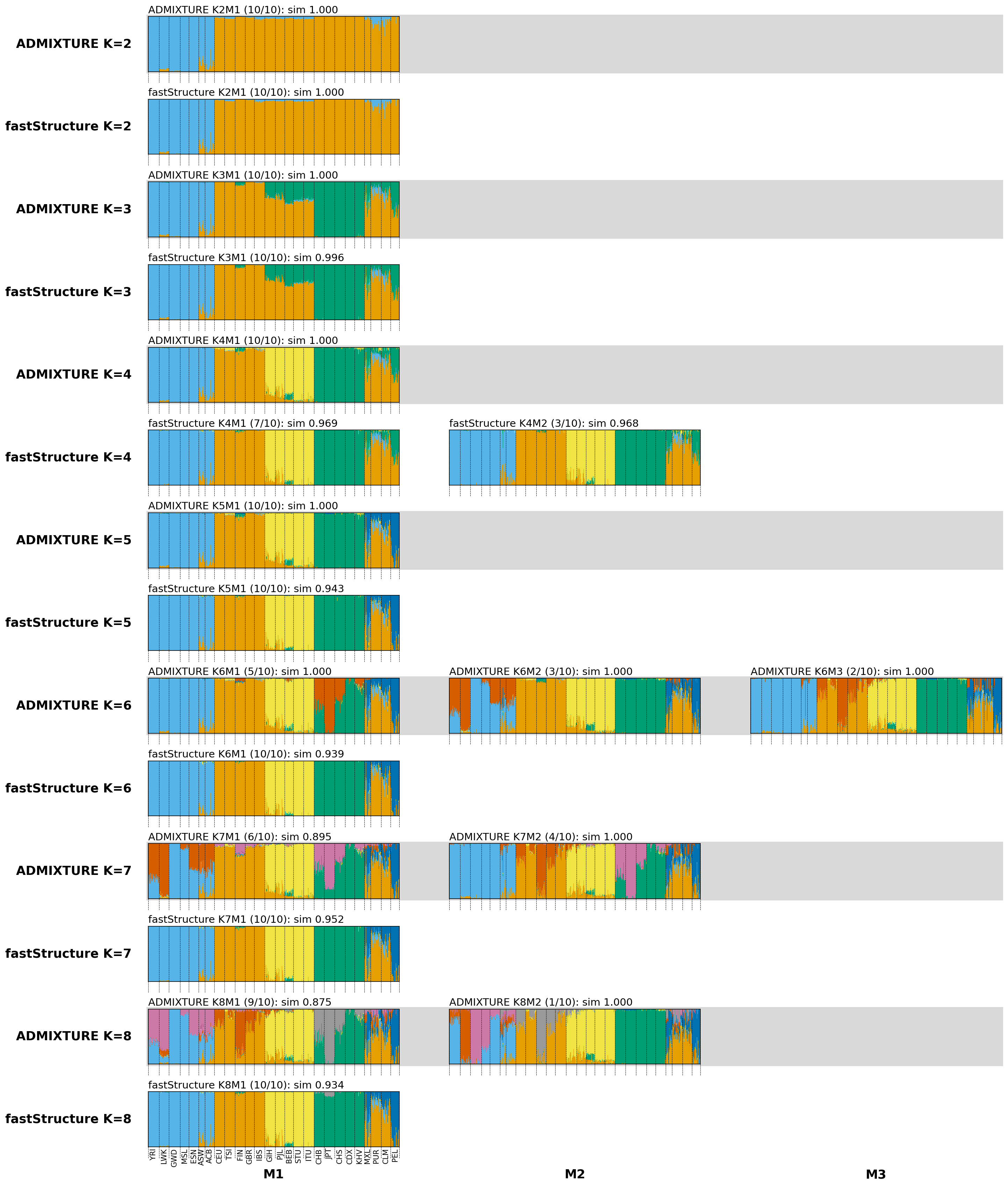
Figure 3. Model comparison of
The Python program
We illustrate the new features of
The alignment pattern graph and bar plots (Figure 2) reveal that at K = 5, multiple plausible explanations exist for the clustering patterns, represented by three modes. The most common mode (K5M1; size=18, similarity=0.82) suggests a new, dominant cluster emerges mainly in Cape Verdean individuals (dark blue). However, an equally frequent mode with lower pairwise alignment similarity (K5M2; size=18, similarity=0.80) indicates a new cluster instead among Iberian and French individuals. A third well-supported mode (K5M3; size=14, similarity=0.85) points to a new cluster separating from the dominant cluster for French and British samples. The presence of these alternative modes indicates that the algorithm is concurrently resolving distinct, fine-scale structure within several groups, and that analyzing a single clustering run at a single value of K is insufficient for interpreting admixture patterns. When multiple modes are identified, we recommend prioritizing modes that are both frequently observed across independent runs (larger size) and internally consistent (higher within-mode alignment similarity). These criteria are complementary: larger mode size often reflects greater support, while within-mode similarity captures how coherently the runs in a mode agree with one another. In practice, we prioritize more frequently occurring modes, while flagging rare but highly consistent modes as potentially meaningful alternatives that may reflect distinct local optima or biologically relevant substructures.
We also performed a comparative analysis of
The results from the two methods are similar for K = 2 to 5, but they diverge at higher K values.
We have introduced
The novel visualization of alignment patterns helps clarify the hierarchical nature of population structure by tracking newly inferred clusters as the number of clusters increases. The new model comparison module allows researchers to evaluate the consistency of results across different analytical choices and helps guide the selection of appropriate data analysis methods. The framework also offers greater algorithmic flexibility and a modular design. These improvements contribute to a clustering alignment framework that can adapt to diverse datasets and methodological choices. The new features facilitate the examination of multimodality, comparing solutions across K and across models, and communicating uncertainty and stability in population-structure inference.
The following supplementary materials are available on the website of this paper: HPGG2606020004SupplementaryMaterials.zip
Appendix A. Population structure analysis of 1000 Genomes Project data.
Figure S1. Clustering results from the 1000 Genomes Project data aligned by Clumppling.
All data used in this study are publicly available either through the cited references or via the program’s GitHub repository at https://github.com/PopGenClustering/Clumppling.
This study was supported by the US National Institutes of Health (NIH) R35 GM139628 to S.R. and R01 HG005855 to N.A.R.
The authors have declared that no competing interests exist.
We thank Cole Williams for inspiring the object-oriented implementation upgrade of the program.
| 1. | Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945-959. [Google Scholar] [CrossRef] |
| 2. | Rosenberg NA. A population-genetic perspective on the similarities and differences among worldwide human populations. Hum Biol. 2011;83(6):659-684. [Google Scholar] [CrossRef] |
| 3. | Lawson DJ, Van Dorp L, Falush D. A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots. Nat Commun. 2018;9(1):3258. [Google Scholar] [CrossRef] |
| 4. | Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164(4):1567-1587. [Google Scholar] [CrossRef] |
| 5. | Corander J, Waldmann P, Marttinen P, Sillanpää MJ. BAPS 2: enhanced possibilities for the analysis of genetic population structure. Bioinformatics. 2004;20(15):2363-2369. [Google Scholar] [CrossRef] |
| 6. | Tang H, Peng J, Wang P, Risch NJ. Estimation of individual admixture: analytical and study design considerations. Genet Epidemiol. 2005;28(4):289-301. [Google Scholar] [CrossRef] |
| 7. | Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655-1664. [Google Scholar] [CrossRef] |
| 8. | Raj A, Stephens M, Pritchard JK. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics. 2014;197(2):573-589. [Google Scholar] [CrossRef] |
| 9. | Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23(14):1801-1806. [Google Scholar] [CrossRef] |
| 10. |
Kopelman NM, Mayzel J, Jakobsson M, et al. Clumpak: a program for identifying clustering modes and packaging population structure inferences across |
| 11. | Behr AA, Liu KZ, Liu-Fang G, et al. Pong: Fast analysis and visualization of latent clusters in population genetic data. Bioinformatics. 2016;32(18):2817-2823. [Google Scholar] [CrossRef] |
| 12. | Liu X, Kopelman NM, Rosenberg NA. A Dirichlet model of alignment cost in mixed-membership unsupervised clustering. J Comput Graph Stat. 2023;32(3):1145-1159. [Google Scholar] [CrossRef] |
| 13. | Liu X, Kopelman NM, Rosenberg NA. Clumppling: cluster matching and permutation program with integer linear programming. Bioinformatics. 2024;40(1):btad751. [Google Scholar] [CrossRef] |
| 14. | Blondel VD, Guillaume JL, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J Stat Mech Theory Exp. 2008. [Google Scholar] [CrossRef] |
| 15. | Van Dongen S. Graph clustering via a discrete uncoupling process. SIAM J Matrix Anal Appl. 2008;30(1):121-141. [Google Scholar] [CrossRef] |
| 16. | 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526(7571):68. [Google Scholar] [CrossRef] |
| 17. | Verdu P, Jewett EM, Pemberton TJ, Rosenberg NA, Baptista M. Parallel trajectories of genetic and linguistic admixture in a genetically admixed creole population. Curr Biol. 2017;27(16):2529-2535. [Google Scholar] [CrossRef] |

![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy




