
Human Population Genetics and Genomics ISSN 2770-5005
Human Population Genetics and Genomics 2023;3(4):0008 | https://doi.org/10.47248/hpgg2303040008
Original Research Open Access
An ancient genome perspective on the dynamic history of the prehistoric Jomon people in and around the Japanese archipelago
Gichan Jeong
†

 ,
Haechan Gill
†
,
Hyungmin Moon
,
Choongwon Jeong
,
Haechan Gill
†
,
Hyungmin Moon
,
Choongwon Jeong
Correspondence: Choongwon Jeong
Academic Editor(s): Joshua Akey
Received: Oct 8, 2023 | Accepted: Dec 1, 2023 | Published: Dec 11, 2023
This article belongs to the Special Issue Paleogenomics, ancient DNA, and genomic tales of human history
© 2023 by the author(s). This is an Open Access article distributed under the Creative Commons License Attribution 4.0 International (CC BY 4.0) license, which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is correctly credited.
Cite this article: Jeong G, Gill H, Moon H, Jeong C. An ancient genome perspective on the dynamic history of the prehistoric Jomon people in and around the Japanese archipelago. Hum Popul Genet Genom 2023; 3(4):0008. https://doi.org/10.47248/hpgg2303040008
The Jomon people were prehistoric residents of the Japanese archipelago who occupied the region from ca. 16,500 to 2,300 years before present (BP). While recent accumulation of ancient genomes and genome-wide data of the Jomons has substantially enhanced our understanding of their genetic profiles and contribution to present-day populations, their genetic history in the Jomon-period archipelago, spanning over 14,000 years in time and 2,000 km in distance, remains scarcely investigated. Here we report multiple findings illuminating the Jomon genetic history based on the analysis of the genetic relationship between published ancient genome-wide data of 23 Jomon and Jomon-like individuals. First, the Initial Jomon individual from Shikoku, dated to ca. 9,000 BP, forms a common outgroup to the remaining later Jomon individuals, suggesting a population turnover in western Japan. Second, genetically Jomon-like individuals outside the Jomon archaeological context, found in the Miyako Island in Ryukyu and the Yokjido island in the southern coast of Korea, show the closest genetic affinity with the Late Jomon individual from Shikoku, narrowing down their sources in space and time. This study highlights a dynamic history of the Jomon people in and out of the Japanese archipelago and calls for a large-scale investigation of the ancient Jomon genomes.
KeywordsJomon, population structure, admixture, Japanese prehistory, maritime migration
The Jomon people are a group that inhabited the Japanese archipelago from approximately 16,500 to 2,300 years before present (BP) [1]. Although predominantly relied on hunting, gathering, and fishing in their subsistence strategy, the Jomon people developed a sedentary lifestyle and manufactured and utilized pottery, the cord-markings on which gave the name Jomon [1,2]. Based on temporal changes in their pottery style, the Jomon period has been separated into the Incipient (ca. 16,500–10,500 BP), Initial (ca. 10,500–7,000 BP), Early (ca. 7,000–5,500 BP), Middle (ca. 5,500-4,500 BP), Late (ca. 4,500–3,250 BP), and Final (ca. 3,250–2,500 BP) Jomon periods [3,4]. The origins and genetic history of the Jomons have been of keen academic interest not just to understand the Jomons themselves, but also to understand the genetic diversity of present-day populations in the Japanese archipelago. For example, the Ainu from the northernmost region of the archipelago are genetically closer to the Ryukyuans from the southernmost region than to the mainland Japanese in the middle, because they inherited higher proportions of their ancestry from the Jomon-related ancestors than the mainland Japanese did [5–12]. Most archaeogenetic studies on the Jomons mainly targeted mitochondrial haplogroups, finding that distinct haplogroups N9b1 and M7a were representative of the northern and southern Jomons, respectively, thus suggesting an internal population differentiation among the Jomons [13–16]. For the last few years, finally ancient genomes or genome-wide data of the Jomons have been reported, providing a chance to investigate the population structure and the genetic history of the Jomons in a fine resolution [17–21]. Particularly, a recent study reported genomes of nine Jomon individuals covering a time period of five millennia (ca. 8,800–3,800 BP) and confirmed a long-standing homogeneous Jomon genetic profile [19].
The recently published Jomon genomes drove substantial progress in major questions about their origins and genetic legacy in present-day populations. For example, the Jomon gene pool is distinct from neighboring populations from the continent because they had a strong genetic link with a deeply branching Eastern Eurasian lineage not well represented by any present-day population [17,22]. The isolation between the Jomon and other East Asian populations was dated to ca. 20–25 thousand years ago (kya) with a small effective population size (<1,000) of Jomon [19]. Also, the tripartite model of Japanese origin, an extension of the traditional dual-structure model [23], has been recently proposed based on the genetic comparison of the ancient Jomon, Yayoi, and Kofun-period individuals [19]. Interestingly, archaeogenetic studies have reported ancient individuals genetically related to the Jomons but found outside the range of the Jomon archaeological culture and out of the Jomon archaeological context [15,20]. This includes the southern islands of the Ryukyu (e.g., Miyako and Yaeyama) and the southern coast of the Korean peninsula. However, these studies have largely focused on the shared common origin of the Jomons and a comparison between the Jomons and the non-Jomons, while leaving the population history within the Jomons largely unexplored.
In this study, we conducted an in-depth re-analysis of the published genome or genome-wide data of the Jomon-related individuals, focusing on the relationship among them. These individuals, 23 in total and ranging between the Initial and Late Jomon periods, provide evidence for rich dynamics within the overall homogeneous Jomon gene pool. Specifically, we found that the Initial Jomon individual from Shikoku forms an outgroup to the later Jomon individuals across the archipelago, suggesting a population turnover in the region. We could also localize the origins of the genetically Jomon-like individuals outside the traditional Jomon area into the Middle/Late Jomon-period western Japan (including Chugoku, Kansai, Shikoku, and Kyushu).
We compiled whole genome or genome-wide sequencing data reported in previous publications, focusing on ancient Jomon and Jomon-related individuals [17–21]. We obtained genome-wide genotype data of these individuals on a set of 1,233,013 ancestry-informative single nucleotide polymorphisms (SNPs) referred to as the “1240K” panel [24,25]. For the studies that made pseudo-haploid pulldown genotype data for the 1240K panel publicly available, we took the published genotype data to guarantee maximum reproducibility. For those that did not release the genotype data, we started from raw sequence data (FASTQ format) or from aligned reads (BAM format), obtained from public sequencing data repositories such as the European Nucleotide Archive (ENA) and the DNA Data Bank of Japan (DDBJ). We provide details of data acquisition information in a separate table (Table S1).
For raw data in the FASTQ format, we first trimmed Illumina adapter sequences from raw reads and merged read pairs for paired-end sequencing data using AdapterRemoval v2.3.0 [26] with a minimum overlap of 1 base pair (bp). We removed trimmed/merged reads shorter than 35 bp and unmerged read pairs from the analysis. We mapped the remaining trimmed/merged reads to the human reference genome with decoy sequences (hs37d5) using the bwa-aln and bwa-samse modules in BWA (Burrows-Wheeler Aligner) v0.7.17 [27]. Then, we removed PCR duplicates using DeDup v0.12.5 [28] and removed reads with the Phred-scaled mapping quality score lower than 30 using SAMtools v1.9 [29].
As ancient DNA has post-mortem DNA damage, we used mapDamage v2.2.1 [30] to check if each individual shows a sufficient level of post-mortem damage matching the pattern expected from the corresponding library preparation method. To minimize the effect of chemical DNA damages in later population genetic analysis, we masked 3 and 10 bps at both ends of reads for partial-UDG and non-UDG treated double-stranded libraries, respectively, using the trimBam function of bamUtil v.1.0.15 [31]. For single-stranded libraries, we did not mask the read ends.
We called pseudo-haploid genotypes using the pileupCaller v1.4.0.5 (https://github.com/stschiff/sequenceTools; v1.5.2 last accessed on 19 April 2023) with the randomHaploid option. For double-stranded libraries, we used masked and non-masked BAM files for calling genotypes of transition and transversion SNPs, respectively. For single-stranded libraries, we used non-masked BAM files and the ‘singleStrandMode’ option implemented in pileupCaller. We estimated relatedness between the Jomon-related individuals based on the pairwise genotype mismatch rate [32] calculated over the autosomal SNPs in the 1240K panel.
We integrated the 1240K genotype data of ancient Jomon individuals [17–21] with two present-day individual datasets. For the 1240K SNP set, we retrieved genotype data of high-coverage whole genome sequenced individuals included in the Simons Genome Diversity Project [33]. For the “HumanOrigins” data set, we took publicly available genome-wide data of a broader set of present-day individuals genotyped on the Affymetrix Axiom Genome-wide Human Origins 1 array ([34–37]). The HumanOrigins array included 593,124 autosomal SNPs overlapping with the 1240K set. We utilized the HumanOrigins dataset for principal component analysis (PCA). To perform f4 symmetry tests between the Jomon individuals against the world-wide population, we employed 1240K genotype data to utilize the genotype information of the Jomon individuals to the maximum degree. The details of the present-day individuals included in this dataset are provided in Table S2.
We performed PCA using the smartpca v18140 from the Eigensoft v8.0.0 package using 2,077 present-day Eurasian individuals genotyped on the HumanOrigins panel [38]. We projected ancient Jomon-related individuals not included in PC calculation using the ‘lsqrproject: YES’ option. The detailed information of present-day individuals used in PCA are provided in Figure S1–S2 and Table S2. For PCA of ancient Jomon-related individuals only, we first computed covariance between each pair of individuals using SNPs not missing in both individuals to avoid an issue due to varying level of genotype missingness across individuals. For this, we used ‘cov’ function and ‘use=“pairwise.complete.obs”’ option implemented in R v4.2.0 and applied the “eigen” function to perform PCA on the calculated covariance matrix.
We calculated the f-statistics by qp3pop and f4 functions from the R library ADMIXTOOLS2 v2.0.0. (https://github.com/uqrmaie1/admixtools, publication pending). We calculated outgroup-f3 using the central African rainforest hunter-gatherer population Mbuti in Congo as an outgroup to measure shared genetic drift between two target populations. We also used Mbuti as an outgroup when calculating f4 statistics. To figure out the population structure within the Jomon-related groups, we calculated all pairs of f4(Mbuti, Jomon 1; Jomon 2, Jomon 3). We calculated f4(Mbuti, worldwide; Jomon 1, Jomon 2) for testing genetic symmetry between Jomon groups or searching for additional admixture sources over worldwide populations. Present-day worldwide populations used in the analysis are listed in Table S2. We calculated standard error measure (s.e.m.) of f3 and f4 statistics using the default 5 cM block jackknife resampling approach.
We investigated runs of homozygosity (ROH) segments of Jomon individuals covering more than 400,000 SNPs among the 1240K panel using hapROH [39]. We used pseudo-haploid genotype call of each Jomon genome and the 1000 Genomes Project Phase 3 haplotype data set as the reference panel [40].
For our study, we used genome-wide data of 23 published ancient Jomon or Jomon-like individuals (Figure 1, Table 1 and Table S1). Among these 23 individuals, 17 are Jomon individuals from the Honshu or Shikoku islands in Japan directly associated with the Jomon archaeological contexts. The remaining 6 harbor a Jomon-like genetic profile but are from an archaeological context not directly related to the Jomons: five are from the Nagabaka site in the Miyako Island of the Ryukyu (“Nagabaka_2800BP” and “Nagabaka_4000BP”), and one is from the Yokjido shell midden site in Tongyeong city, Republic of Korea (“Yokjido_4000BP”). We detected no close relatives among the 23 individuals and therefore included all of them in the analysis (Figure S3 and Table S3). We split them into 11 analysis groups based on their archaeological site and date information (Figure 1, Table 1 and Table S1). Among the individuals used, we flagged two individuals for low quality: the Yokjido individual (TYJ001) was potentially contaminated to a small degree (6% contamination based on its mitochondrial capture data) and the oldest Nagabaka individual (NAG016; Nagabaka_4000BP) was heavily contaminated and of low coverage (27,652 out of 1,233,013 SNPs in the 1240K panel were covered). Because the unknown contaminants are a priori unlikely to be of the Jomon origin, which no longer exists among the present-day individuals, we included them in a subset of the analyses where contamination would not affect the tests in a qualitative manner. We also used four additional prehistoric individuals from the southern coast of Korea who were reported to have a Jomon genetic affinity: two from the Janghang Neolithic cemetery site in Busan city (“Janghang_6700BP”) and the other two from the Yeondae-do shell midden site in Tongyeong city (“Yeondaedo_7000BP”) [20].

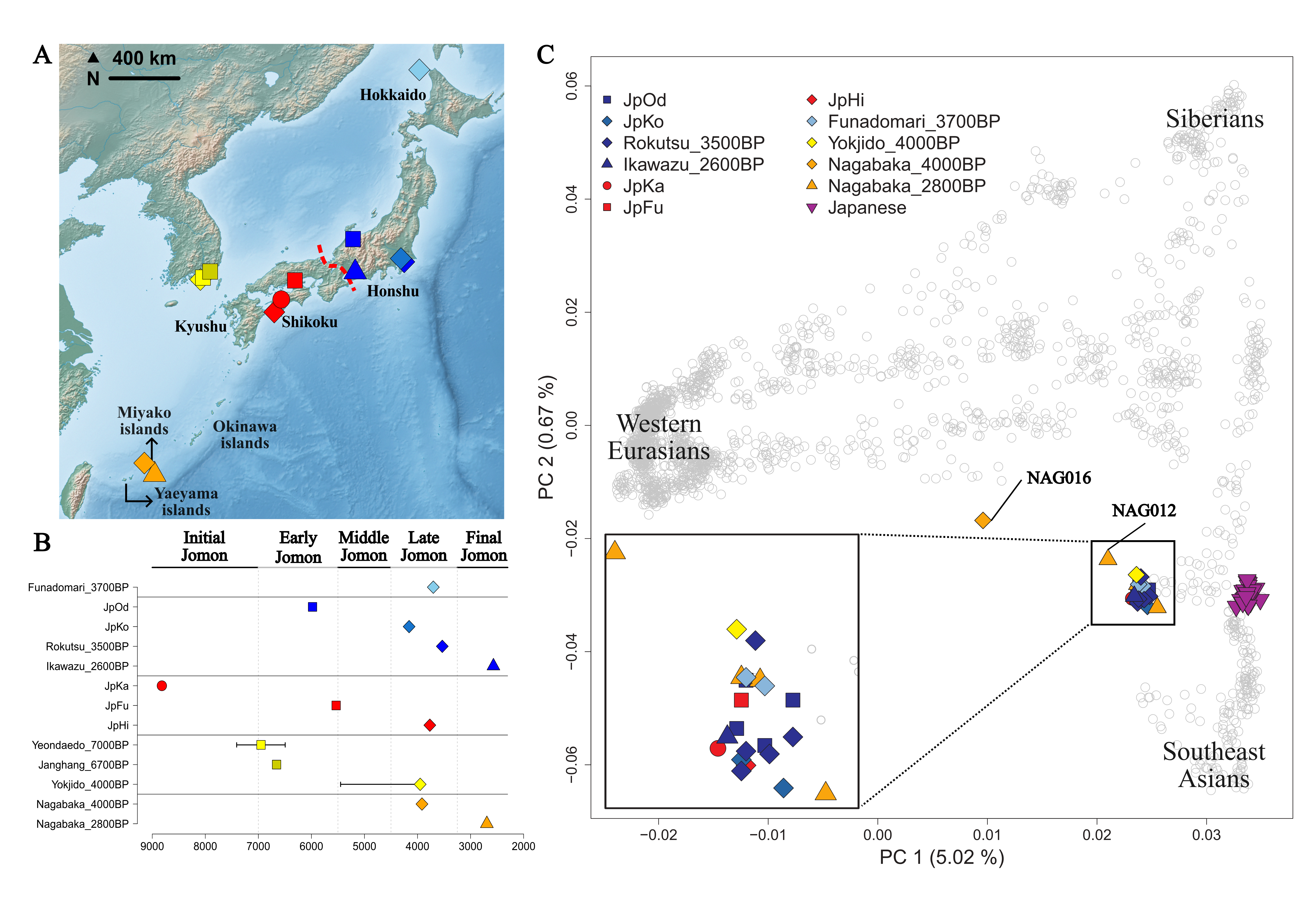
Figure 1 Ancient Jomon-related groups used in this study. (A) Geographical location of the sites of each Jomon-related group used in this study. The red dotted line in Honshu marks the boundary between western and eastern Japan. Site names are listed in (B). (B) The average estimated dates of each Jomon-related group are represented as color-filled shapes. For groups not directly radiocarbon dated, the reported lower and upper bounds based on archaeological contexts are represented as the horizontal bars. The base map was obtained from the Natural Earth public domain map data set (https://www.naturalearthdata.com/download/10m/raster/HYP_LR_SR_OB_DR.zip) and modified using R v4.2.0. (C) Jomon-related individuals (color-filled shapes other than present-day Japanese) were projected onto the top two PCs calculated with 2,077 present-day Eurasians (gray circles). Among present-day Eurasians, we highlighted present-day mainland Japanese with purple-filled downward triangles. A zoom-in plot of the Jomon cluster is shown in the bottom-left. Population labels and the geographic locations of present-day Eurasian individuals are presented in Figures S1-S2 and Table S2.
Table 1 List of Jomon-related individuals used in this study. This list includes meta information of 27 Jomon-related individuals used in this study. Date information represent the 2-sigma range of the calibrated dates for directly radiocarbon-dated individuals and the context-based estimate for the others. More detailed metadata of the 27 Jomon-related individuals is provided in Table S1.
We performed principal components analysis (PCA) to make a visual summary of the genetic profile of the Jomon-related individuals in comparison to present-day Eurasian populations (Figure 1C). Consistent with previous studies reporting a homogeneous genetic profile across the Jomon-related individuals [19], the Jomon-related individuals form a tight and distinct cluster separated from other populations. Two individuals deviating from the cluster are from Nagabaka (NAG012 and NAG016): both with low coverage and one with heavy contamination (NAG016).
To investigate a fine-scaled genetic stratification within the Jomon-related individuals, we performed PCA exclusively with Jomon, Yokjido, and Nagabaka individuals. In this analysis we observe a separation of Nagabaka and Yokjido individuals from the other Jomon individuals (Figure S4A). When we apply PCA only to the Jomon individuals in the mainland Japan, excluding Nagabaka and Yokjido, we observe a separation between Jomons from western Japan, eastern Japan (Chubu and Kanto), and Hokkaido (Figure S4B).
Considering the low coverage nature of most Jomon-related individuals, we measured the level of genetic diversity within each Jomon-related group by calculating pairwise genotype mismatch rate (PMR) between individuals (Figure S3 and Table S3). PMR values between individuals from different Jomon groups (mean = 0.191) are overall similar and much lower than those from present-day East Asians (mean = 0.241), such as Han Chinese and Japanese, suggesting overall reduced genetic diversity within the Jomon metapopulation. Interestingly, Nagabaka_2800BP individuals show even more reduced PMR (mean = 0.154), implying a strong population bottleneck specific to Nagabaka. The distribution of runs of homozygosity (ROH) segments provides a similar pattern (Figure S5). That is, the Jomon-related individuals overall show an accumulation of ROH segments, a signature of reduced genetic diversity, and Nagabaka individuals tend to have more ROH segments than most other Jomon individuals.
To investigate the population structure among the Jomon-related groups, we first measured the genetic affinity between each pair of the Jomon-related groups using the outgroup-f3 statistic in the form of f3(Mbuti; Jomon 1, Jomon 2) (Figures S6–S7 and Table S4). We used Mbuti, a Central African rainforest hunter-gatherer population, as an outgroup and applied the analysis to 9 out of 11 Jomon-related groups, excluding Nagabaka_4000BP and Yokjido_4000BP due to contamination. When included, they showed consistently lower outgroup-f3 values than other pairs (Figure S6A). We find that the Initial Jomon individual from Shikoku (JpKa; dated to ca. 9,000 BP) shows a relatively low genetic affinity with other Jomons (Figure S6B). Among all pairs, the pair of an Early Jomon individual from western Honshu (JpFu, dated to ca. 5,600 BP) and a Late Jomon individual from Shikoku (JpHi, dated to ca. 3,800 BP) shows the highest value (Figure S7 and Table S4).
Inspired by the outgroup-f3 results and the presence of the Initial Jomon individual, we explicitly compared two competing hypotheses regarding the population structure of the Jomons: (1) the oldest Jomon individual JpKa forms a common outgroup to all of the later Jomon individuals, and (2) the Jomon population in western Japan (JpKa, JpFu, JpHi) shows a population continuity over a 5,000-year-period (Figure 2). To distinguish between the two hypotheses, we calculated f4 statistics in the form of f4(Mbuti, Jomon 1; Jomon 2, Jomon 3) for all triplets of the Jomon-related groups (Figure 3, Figures S8-S9 and Table S5). Under the former hypothesis, f4(Mbuti, JpKa; Jomon 2, Jomon 3) is expected to be zero for all (Jomon 2, Jomon 3) pairs because JpKa is a common outgroup to all later Jomon groups. In contrast, under the latter hypothesis, JpKa is supposed to be genetically closer to later Western Jomon groups (JpFu/JpHi) than to the other Jomons, thus we predict f4(Mbuti, JpKa; Jomon 2, Jomon 3) to be significantly positive when Jomon 3 is later Jomon populations from western Japan (i.e., JpFu and JpHi). Likewise, f4(Mbuti, JpFu/JpHi; JpKa, Jomon 3) will be positive under the former hypothesis while it will be negative under the latter.
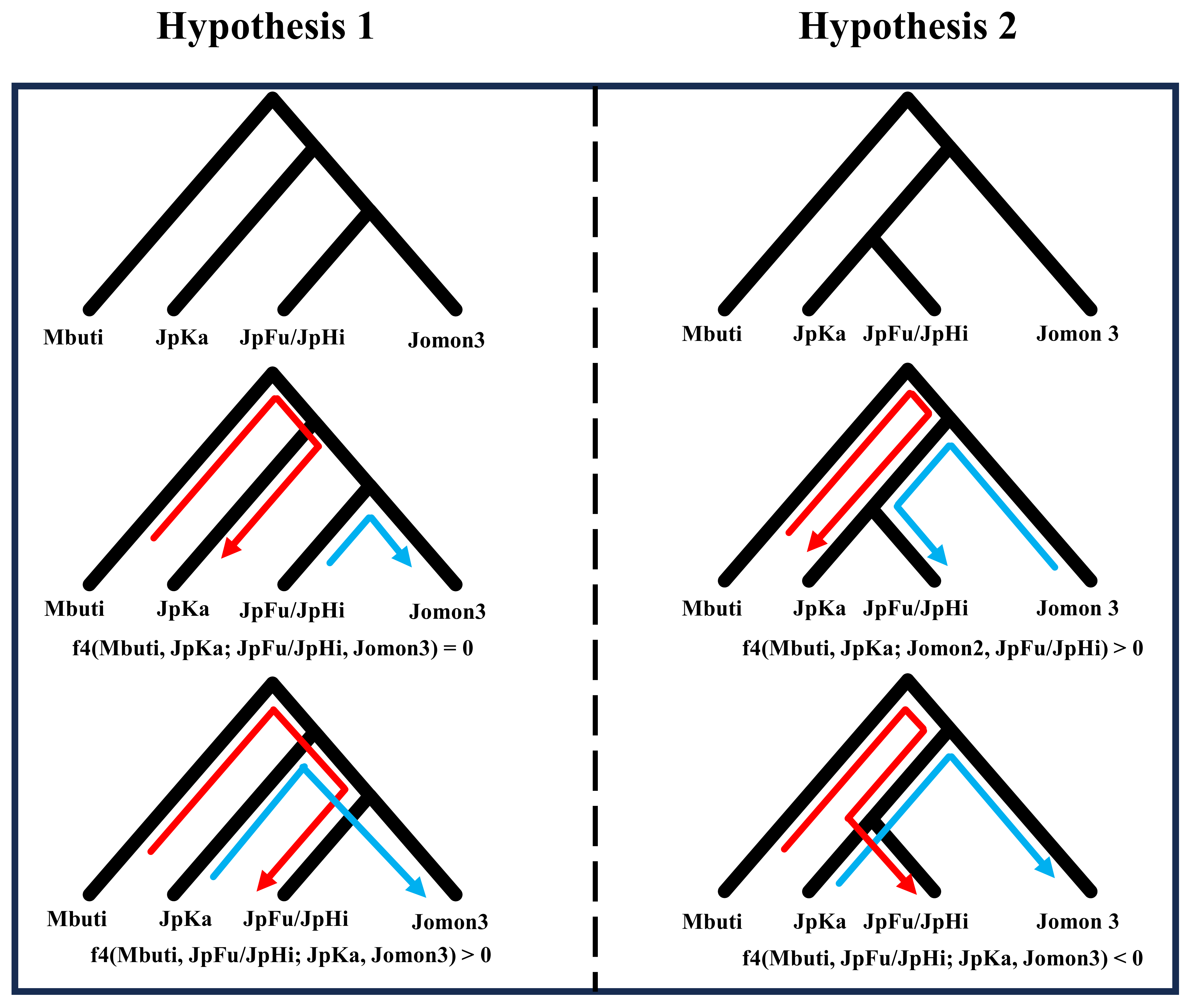
Figure 2 Two competing hypotheses regarding population structure of the Jomon. We suggest two competing hypotheses with f4 statistics the expection of which can distinguish between the two. (Left side) Hypothesis 1 assumes that the oldest Jomon from western Japan, JpKa, is a common outgroup to later Jomons including those from the nearby region (JpFu/JpHi). (Right side) Hypothesis 2 assumes a genetic continuity in western Japan, suggesting that the older JpKa and later JpFu/JpHi form a clade against those from eastern Japan. Red and skyblue arrows represent drift paths from population 1 to 2 (red) and population 3 to 4 (skyblue) in f4(population 1, population 2; population 3, population 4), respectively.
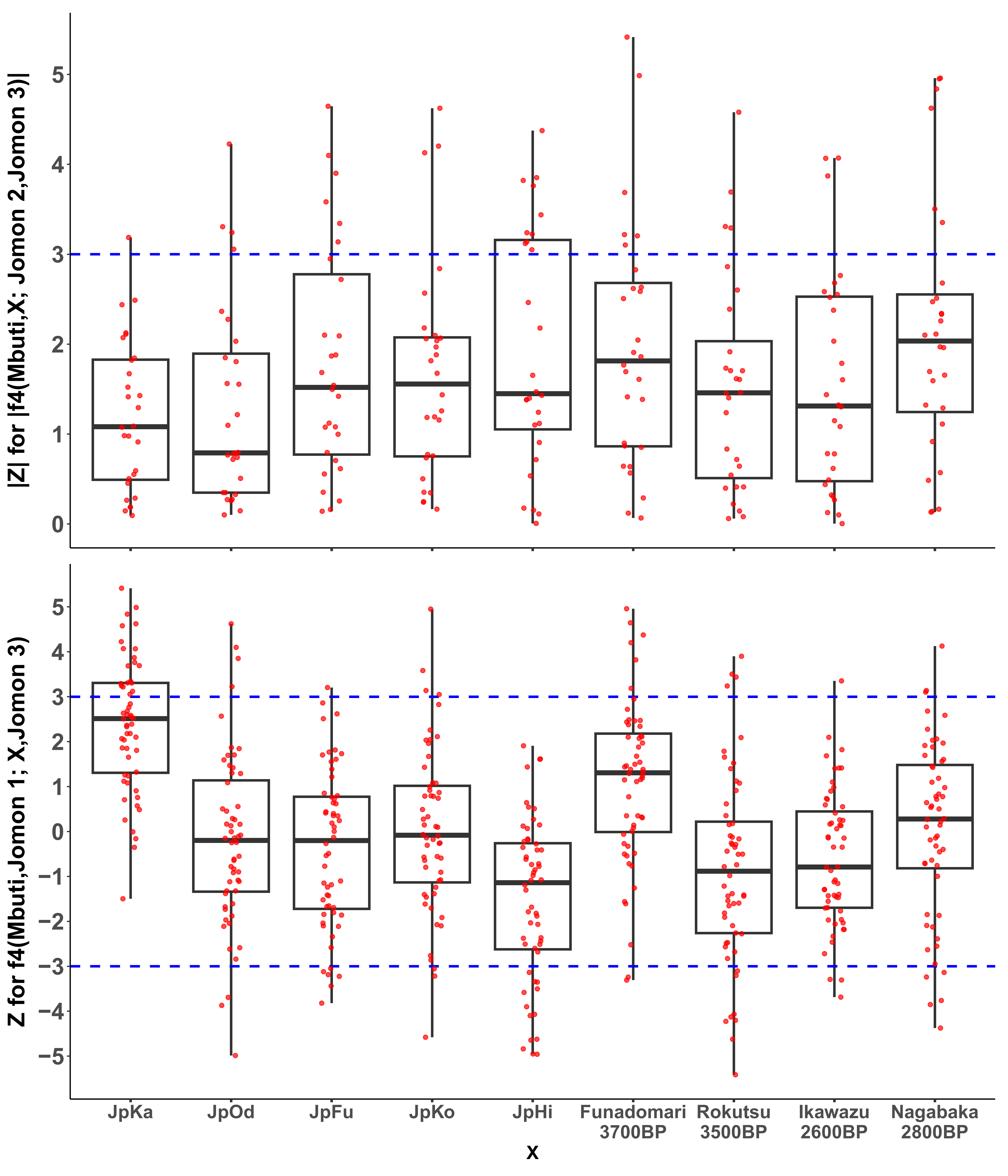
Figure 3 Cladality test for all pairs of Jomon individuals. We calculated f4 statistics between the Jomon groups to identify the most distantly related one among them (i.e., a common outgroup). (A) We present absolute Z-scores of the f4 statistics in the form of f4(Mbuti, X; Jomon 2, Jomon 3) using a box-whisker plot. Z-scores are calculated by a 5cM block jackknife resampling approach. This statistic is expected to be zero when X represents the common outgroup. (B) We present Z-scores of the f4 statistics in the form of f4(Mbuti, Jomon 1; X, Jomon 3). Positive values indicate an excess shared genetic drift between (Jomon 1, Jomon 3) pairs against X.
For the statistic f4(Mbuti, JpKa; Jomon 2, Jomon 3), only 1 out of the 28 Jomon group pairs has |Z| > 3 value: f4(Mbuti, JpKa; Funadomari_3700BP, JpFu) = 3.187 s.e.m. (Figure 3A, Figure S8 and Table S5). The other eight Jomon groups have 3 to 10 pairs with |Z| > 3 values when they are placed in the second population, making them less likely to be a common outgroup to the other Jomons (Figure 3A, Figure S8 and Table S5). Furthermore, f4(Mbuti, Jomon 1; JpKa, Jomon 3) are significantly positive in many (Jomon 1, Jomon 3) pairs and in general tend to be positive in its value, preferring the former hypothesis over the latter (Figure 3B, Figure S8 and Table S5).
The second statistic, f4(Mbuti, JpFu/JpHi; JpKa, Jomon 3) are mostly |Z| < 3, matching the prediction by neither hypothesis (Table S5). Comparing other Jomon groups’ affinity to JpKa and JpFu/JpHi by f4(Mbuti, Jomon 1; JpKa, JpFu/JpHi), the results are not statistically significant but mostly positive. We speculate that the Early Jomon JpFu may have a partial genetic link with JpKa, the earlier individual from a nearby region, while formally testing this admixture hypothesis may need more ancient genomes to enhance statistical power. Therefore, the oldest Jomon individual, JpKa, is likely to represent a common outgroup to the available Jomon groups, including those from western Japan.
Finally, we applied the same analysis to the second oldest Jomon group from the Early Jomon Odake shell-midden site from north-central Honshu (JpOd; dated to ca. 6,000 BP). We find no Jomon pairs that are asymmetrically related to JpOd by observing f4(Mbuti, JpOd; Jomon 2, Jomon 3) within 3 s.e.m. in all Jomon pairs but those including JpKa: f4(Mbuti, JpOd; JpKa, Jomon 3) > 3 s.e.m. for all later Jomon groups from eastern Japan (including Chubu, Kanto, and Hokkaido) (Table S5).
We investigated possible origins of individuals from the Nagabaka (“Nagabaka_2800BP”) and the Yokjido shell midden sites (“Yokjido_4000BP”), who exhibited a Jomon-like genetic profile, although these sites and regions are not directly associated with the Jomon archaeological culture. We found that JpHi, the Late Jomon from Shikoku, is the closest Jomon group with Nagabaka_2800BP: f4(Mbuti, Nagabaka_2800BP; other Jomon, JpHi) are mostly significantly positive (Z = 2.5 to 4.9) (Figure 4A and Table S5). The older Early Jomon individual from western Honshu, JpFu, is second most closely related to Nagabaka_2800BP although the difference is not statistically significant: f4(Mbuti, Nagabaka_2800BP; other Jomon, JpFu) = 0.1 to 2.3 s.e.m. except for JpHi (Z=-2.5) (Table S5). The Jomon-related population would have been already in Nagabaka by ca. 4,000 BP, considering a clear Jomon affinity of the older Nagabaka individual (Nagabaka_4000BP; directly dated to ca. 4,000 BP) [20]. Taken together, the Late Jomon population from western Japan, related to JpFu/JpHi, was likely a source of the Jomon-related population in southern Ryukyu.
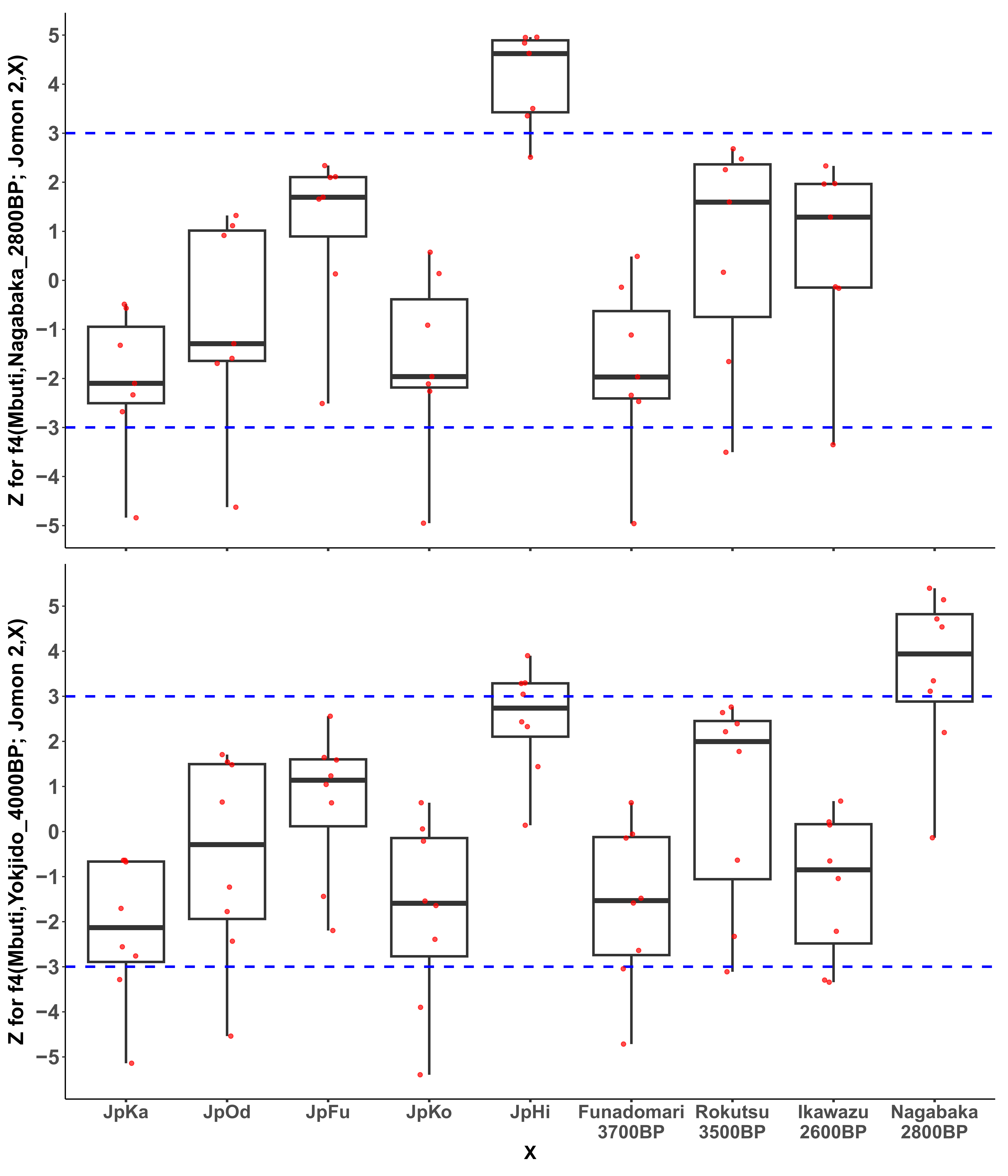
Figure 4 The genetic affinity of Nagabaka_2800BP and Yokjido_4000BP with the Jomon groups. (A) We present the f4 statistics in the form of f4(Mbuti, Nagabaka_2800BP; Jomon 2, X) to find the Jomon groups most closely related to Nagabaka_2800BP. Z-scores of f4 statistics are calculated by a 5cM block jackknife resampling approach and presented by box-whisker plots. (B) We present the f4 statistics in the form of (Mbuti, Yokjido_4000BP; Jomon 2, X) to find the Jomon groups most closely related to Yokjido_4000BP. A positive value obtained from this calculation indicates high genetic affinity between X and the targets (Nagabaka_2800BP and Yokjido_4000BP) against other Jomon groups.
Likewise, we find that JpHi and Nagabaka_2800BP are the closest Jomon-related group with Yokjido_4000BP: f4(Mbuti, Yokjido_4000BP; Jomon, JpHi/Nagabaka_2800BP) = 2.3 to 5.4 s.e.m. and f4(Mbuti, Yokjido_4000BP; Nagabaka_2800BP, JpHi) = 0.1 s.e.m. (Figure 4B and Table S6). A previous study reported the Jomon ancestry contribution in prehistoric groups along the southern coast of Korea: Yeondaedo_7000BP and Janghang_6700BP [20]. Repeating the same analysis, we find that JpHi seems to be the closest Jomon-related group with them: f4(Mbuti, Yeondaedo_7000BP; other Jomon, JpHi) = 0.0 to 1.6 s.e.m and f4(Mbuti, Janghang_6700BP; other Jomon, JpHi) = 1.2 to 2.7 s.e.m., but none of the tests are statistically significant (i.e., Z < 3) due to small sample size and low coverage (Table S6). To summarize, we suggest that the dispersal of the Jomon-related populations into southern Korea and the Ryukyu islands, clearly observed by ca. 4,000 and 2,800 BP in the two regions, respectively, is likely to have originated from western Japan, in agreement with the geographic proximity of these regions. While it is possible that the Jomon ancestry found in the earlier individuals in these regions (ca. 6,500–7,000 BP in southern Korea (Janghang_6700BP and Yeondaedo_7000BP) and ca. 4,000 BP in Miyako) may have been derived from the same source, we caution that the currently available genome data lack a resolution to distinguish between different Jomon sources. Future ancient genome sampling over these regions, as well as across the Japanese archipelago, will help us narrow down the time window and true sources of the Jomon ancestry in these regions.
The origins and history of the Jomon people, who had occupied the Japanese archipelago for more than 14,000 years, have long been investigated by archaeologists and geneticists. Recent advances in archaeogenomics have finally made a major breakthrough by decoding dozens of ancient Jomon genomes [17–21]. Although these studies clarified a distinct genetic origin of the Jomons and their overall homogeneous genetic profiles, they focused less on reconstructing the genetic diversity and relationship among the Jomons. Here we investigate the Jomon population structure over time in a comprehensive manner based on a compilation of the Jomon genomes so far available.
We find that the Initial Jomon individual from Shikoku, JpKa, forms an outgroup common to later Jomon groups in both western and eastern Japan, while having a residual affinity with later groups in the nearby regions in western Japan. The only pair of Jomon groups that breaks symmetry, JpFu and Funadomari, may be due to this residual affinity between JpKa and JpFu. Alternatively, this may be due to Funadomari-specific genetic connection, i.e., a link around the Sea of Okhotsk, suggested by the observation that Asiatic Eskimos and Ulchi from the Sakhalin Island, among the present-day worldwide populations, are closer to Funadomari than to JpKa, by the f4 statistic of the form f4(Mbuti, worldwide; Funadomari, JpKa) (Table S7). A previous study reported a similar connection between the Ainu from Hokkaido and the populations around the Sea of Okhotsk, although this signal may be due to a more recent genetic exchange between them [12]. The next oldest Jomon group, the Early Jomon JpOd, in contrast shows a clear genetic affinity with all later Jomons from eastern Japan. A parsimonious scenario is that the eastern Jomon population expanded into the west and partially replaced the local western population at some point within a broad time range of ca. 9,000 and 5,500 BP (i.e., between JpKa and JpFu). We caution that due to sparse sampling of the Jomon genomes over space and time the representativeness of our findings may not be guaranteed.
While the Jomons have been frequently assumed to be strongly isolated from the populations outside the Japanese archipelago, recent archaeological studies highlight prolonged cultural contacts with continental populations prior to their major contact with the Yayoi people in the late first millennium BCE [41–43]. These studies reported the appearance of material cultural elements, such as domesticated plants (e.g., azuki and soybeans) and bronze objects, from the continent to the Jomon-period Japan. Interestingly, the recent archaeogenomic studies reported a Jomon-like genetic profile in ancient individuals outside the Jomon archaeological context, including the Miyako island in the southern Ryukyu and the Yokjido site in the southern coast of Korea [20]. The Jomon-like individuals in both sites harbor the strongest genetic affinity with the Late Jomon individual from western Japan (JpHi; dated to ca. 3,800 BP). In both regions, the earliest appearance of the Jomon ancestry is far older than the time of JpHi while older individuals there are of low quality and thus unable to pinpoint the affinity to specific Jomon groups: ca. 4,000 BP in Miyako (Nagabaka_4000BP) and ca. 6,500–7,000 BP in southern Korea (Janghang_6700BP and Yeondaedo_7000BP). Therefore, the spatial and temporal origins of the Jomon populations who contributed to these early populations remain obscure. Further sampling of the Jomon genomes, especially the Early/Middle Jomons from Kyushu, the westernmost major island in Japan, will provide a pivotal piece of information to reconstruct the outbound genetic connection of the Jomons with the Eurasian continent.
The following supplementary materials can be downloaded at: HPGG2303040008SupplementaryMaterials.zip.
Figure S1. Present-day Eurasian populations used in principal component analysis.
Figure S2. Geographic location of present-day Eurasian populations used in principal component analysis.
Figure S3. Pairwise genotype mismatch rate within and between the Jomon-related groups.
Figure S4. PCA results of the Jomon-related individuals.
Figure S5. Small effective population size of the Jomons shown by runs of homozygosity segments.
Figure S6. Heatmap of outgroup-f3 statistics of the Jomon-related groups.
Figure S7. Outgroup-f3 statistics of Jomon-related groups.
Figure S8. Genetic symmetry test between Jomon-related groups.
Figure S9. Genetic affinity test of Jomon-related groups.
Table S1. Meta information of Jomon-related individuals used in this study.
Table S2. List of present-day populations and non-Jomon ancient population used in this study.
Table S3. Pairwise mismatch rate between Jomon Individuals.
Table S4. The genetic affinity between Jomon groups.
Table S5. F4 statistic for all triplets of the Jomon-related groups.
Table S6. F4 statistics for comparing the Jomon group pairs with regard to their genetic affinity to the Jomon-related groups outside the Jomon archaeological contexts.
Table S7. The relative genetic affinity between JpKa and other Jomon-related groups to present-day world-wide populations.
Not applicable.
Not applicable.
The genotype data for the 1240K panel have been deposited in the Edmond Data Repository of the Max Planck Society [https://edmond.mpg.de/dataset.xhtml?persistentId=doi:10.17617/3.CXKGCX]. All analyses performed in this study are based on publicly available programs. Program names, versions, and non-default options are described in detail in the Methods section. All scripts used for the analyses presented in this study are publicly available via the Github repository (https://github.com/CWJeongLab/HPGG_Jomon).
This work was supported by National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT): RS-2023-00212640 (C.J.).
The authors have declared that no competing interests exist.
Conceptualization: CJ; Methodology: GJ, HG, HM, CJ; Investigation: GJ, HG; Visualization: GJ, HG, HM; Supervision: CJ; Writing—original draft: GJ, HG; Writing—review & editing: GJ, HG, HM, CJ.
| 1. | Habu J. Ancient Jomon of Japan. Cambridge: Cambridge University Press; 2004. |
| 2. | Craig OE, Saul H, Lucquin A, Nishida Y, Taché K, Clarke L, Thompson A, Altoft DT, Uchiyama J, Ajimoto M, Heron CP, Jordan P. Earliest evidence for the use of pottery. Nature. 2013;496:351-354. [Google Scholar] [CrossRef] |
| 3. | Kobayashi T, Hudson M, Yamagata M. Regional organization in the Jomon period. Arctic Anthropol. 1992;29:82-95. [Google Scholar] |
| 4. | Sakaguchi T. Storage adaptations among hunter–gatherers: A quantitative approach to the Jomon period. J Anthropol Archaeol. 2009;28:290-303. [Google Scholar] [CrossRef] |
| 5. | Tajima A, Pan IH, Fucharoen G, Fucharoen S, Matsuo M, Tokunaga K, Juji T, Hayami M, Omoto K, Horai S. Three major lineages of Asian Y chromosomes: implications for the peopling of east and southeast Asia. Hum Genet. 2002;110:80-88. [Google Scholar] [CrossRef] |
| 6. | Matsukusa H, Oota H, Haneji K, Toma T, Kawamura S, Ishida H. A genetic analysis of the Sakishima islanders reveals no relationship with Taiwan aborigines but shared ancestry with Ainu and main-island Japanese. Am J Phys Anthropol. 2010;142:211-223. [Google Scholar] [CrossRef] |
| 7. | Koganebuchi K, Katsumura T, Nakagome S, Ishida H, Kawamura S, Oota H. Autosomal and Y-chromosomal STR markers reveal a close relationship between Hokkaido Ainu and Ryukyu islanders. Anthropol Sci. 2012;120:199-208. [Google Scholar] [CrossRef] |
| 8. | Nishida N, Hirai M, Kawamura S, Oota H, Umetsu K, Kimura R, Ohashi J, Tajima A, Yamamoto T, Tanabe H, Mano S, Suto Y, Omoto K, Tokunaga K, Saitou N. The history of human populations in the Japanese Archipelago inferred from genome-wide SNP data with a special reference to the Ainu and the Ryukyuan populations. J Hum Genet. 2012;57:787-795. [Google Scholar] [CrossRef] |
| 9. | Sato T, Nakagome S, Watanabe C, Yamaguchi K, Kawaguchi A, Koganebuchi K, Haneji K, Yamaguchi T, Hanihara T, Yamamoto K, Ishida H, Mano S, Kimura R, Oota H. Genome-wide SNP analysis reveals population structure and demographic history of the Ryukyu islanders in the southern part of the Japanese archipelago. Mol Biol Evol. 2014;31:2929-2940. [Google Scholar] [CrossRef] |
| 10. | Jinam TA, Kanzawa-Kiriyama H, Inoue I, Tokunaga K, Omoto K, Saitou N. Unique characteristics of the Ainu population in Northern Japan. J Hum Genet. 2015;60:565-571. [Google Scholar] [CrossRef] |
| 11. | Nakagome S, Sato T, Ishida H, Hanihara T, Yamaguchi T, Kimura R, Mano S, Oota H, Asian DNA Repository Consortium. Model-based verification of hypotheses on the origin of modern Japanese revisited by Bayesian inference based on genome-wide SNP data. Mol Biol Evol. 2015;32:1533-1543. [Google Scholar] [CrossRef] |
| 12. | Jeong C, Nakagome S, Di Rienzo A. Deep history of East Asian populations revealed through genetic analysis of the Ainu. Genetics. 2016;202:261-272. [Google Scholar] [CrossRef] |
| 13. | Kivisild T, Tolk H-V, Parik J, Wang Y, Papiha SS, Bandelt H-J, Villems R. The emerging limbs and twigs of the East Asian mtDNA tree. Mol Biol Evol. 2002;19:1737-1751. [Google Scholar] [CrossRef] |
| 14. | Adachi N, Shinoda Ki, Umetsu K, Kitano T, Matsumura H, Fujiyama R, Sawada J, Tanaka M. Mitochondrial DNA analysis of Hokkaido Jomon skeletons: remnants of archaic maternal lineages at the southwestern edge of former Beringia. Am J Phys Anthropol. 2011;146:346-360. [Google Scholar] [CrossRef] |
| 15. | Shinoda K-i, Adachi N. Ancient DNA analysis of Palaeolithic Ryukyu islanders. Terra Australis. 2017;45:51-59. [Google Scholar] [CrossRef] |
| 16. | Mizuno F, Taniguchi Y, Kondo O, Hayashi M, Kurosaki K, Ueda S. Diversity in matrilineages among the Jomon individuals of Japan. Ann Hum Biol. 2023;50:324-331. [Google Scholar] [CrossRef] |
| 17. | McColl H, Racimo F, Vinner L, Demeter F, Gakuhari T, Moreno-Mayar JV, Van Driem G, Gram Wilken U, Seguin-Orlando A, De la Fuente Castro C, Wasef S, Shoocongdej R, Souksavatdy V, Sayavongkhamdy T, Saidin MM, Allentoft ME, Sato T, Malaspinas A-S, Aghakhanian FA, Korneliussen T, Prohaska A, Margaryan A, De Barros Damgaard P, Kaewsutthi S, Lertrit P, Nguyen TMH, Hung H-c, Minh Tran T, Nghia Truong H, Nguyen GH, Shahidan S, Wiradnyana K, Matsumae H, Shigehara N, Yoneda M, Ishida H, Masuyama T, Yamada Y, Tajima A, Shibata H, Toyoda A, Hanihara T, Nakagome S, Deviese T, Bacon AM, Duringer P, Ponche J-L, Shackelford L, Patole-Edoumba E, Nguyen AT, Bellina-Pryce B, Galipaud J-C, Kinaston R, Buckley H, Pottier C, Rasmussen S, Higham T, Foley RA, Lahr MM, Orlando L, Sikora M, Phipps ME, Oota H, Higham C, Lambert DM, Willerslev E. The prehistoric peopling of Southeast Asia. Science. 2018;361:88-92. [Google Scholar] [CrossRef] |
| 18. | Kanzawa-Kiriyama H, Jinam TA, Kawai Y, Sato T, Hosomichi K, Tajima A, Adachi N, Matsumura H, Kryukov K, Saitou N, Shinoda K-i. Late Jomon male and female genome sequences from the Funadomari site in Hokkaido, Japan. Anthropol Sci. 2019;127:83-108. [Google Scholar] [CrossRef] |
| 19. | Cooke NP, Mattiangeli V, Cassidy LM, Okazaki K, Stokes CA, Onbe S, Hatakeyama S, Machida K, Kasai K, Tomioka N, Matsumoto A, Ito M, Kojima Y, Bradley DG, Gakuhari T, Nakagome S. Ancient genomics reveals tripartite origins of Japanese populations. Sci Adv. 2021;7:eabh2419. [Google Scholar] [CrossRef] |
| 20. | Robbeets M, Bouckaert R, Conte M, Savelyev A, Li T, An D-I, Shinoda K-i, Cui Y, Kawashima T, Kim G, Uchiyama J, Dolińska J, Oskolskaya S, Yamano K-Y, Seguchi N, Tomita H, Takamiya H, Kanzawa-Kiriyama H, Oota H, Ishida H, Kimura R, Sato T, Kim J-H, Deng B, Bjørn R, Rhee S, Ahn K-D, Gruntov I, Mazo O, Bentley JR, Fernandes R, Roberts P, Bausch IR, Gilaizeau L, Yoneda M, Kugai M, Bianco RA, Zhang F, Himmel M, Hudson MJ, Ning C. Triangulation supports agricultural spread of the Transeurasian languages. Nature. 2021;599:616-621. [Google Scholar] [CrossRef] |
| 21. | Wang C-C, Yeh H-Y, Popov AN, Zhang H-Q, Matsumura H, Sirak K, Cheronet O, Kovalev A, Rohland N, Kim AM, Mallick S, Bernardos R, Tumen D, Zhao J, Liu Y-C, Liu J-Y, Mah M, Wang K, Zhang Z, Adamski N, Broomandkhoshbacht N, Callan K, Candilio F, Carlson KSD, Culleton BJ, Eccles L, Freilich S, Keating D, Lawson AM, Mandl K, Michel M, Oppenheimer J, Özdoğan KT, Stewardson K, Wen S, Yan S, Zalzala F, Chuang R, Huang C-J, Looh H, Shiung C-C, Nikitin YG, Tabarev AV, Tishkin AA, Lin S, Sun Z-Y, Wu X-M, Yang T-L, Hu X, Chen L, Du H, Bayarsaikhan J, Mijiddorj E, Erdenebaatar D, Iderkhangai T-O, Myagmar E, Kanzawa-Kiriyama H, Nishino M, Shinoda K-i, Shubina OA, Guo J, Cai W, Deng Q, Kang L, Li D, Li D, Lin R, Nini, Shrestha R, Wang L-X, Wei L, Xie G, Yao H, Zhang M, He G, Yang X, Hu R, Robbeets M, Schiffels S, Kennett DJ, Jin L, Li H, Krause J, Pinhasi R, Reich D. Genomic insights into the formation of human populations in East Asia. Nature. 2021;591:413-419. [Google Scholar] [CrossRef] |
| 22. | Gakuhari T, Nakagome S, Rasmussen S, Allentoft ME, Sato T, Korneliussen T, Chuinneagáin BN, Matsumae H, Koganebuchi K, Schmidt R, Mizushima S, Kondo O, Shigehara N, Yoneda M, Kimura R, Ishida H, Masuyama T, Yamada Y, Tajima A, Shibata H, Toyoda A, Tsurumoto T, Wakebe T, Shitara H, Hanihara T, Willerslev E, Sikora M, Oota H. Ancient Jomon genome sequence analysis sheds light on migration patterns of early East Asian populations. Commun Biol. 2020;3:437. [Google Scholar] [CrossRef] |
| 23. | Hanihara K. Dual structure model for the population history of the Japanese. Japan Rev. 1991;2:1-33. [Google Scholar] |
| 24. | Mathieson I, Lazaridis I, Rohland N, Mallick S, Patterson N, Roodenberg SA, Harney E, Stewardson K, Fernandes D, Novak M, Sirak K, Gamba C, Jones ER, Llamas B, Dryomov S, Pickrell J, Arsuaga JL, de Castro JMB, Carbonell E, Gerritsen F, Khokhlov A, Kuznetsov P, Lozano M, Meller H, Mochalov O, Moiseyev V, Guerra MAR, Roodenberg J, Vergès JM, Krause J, Cooper A, Alt KW, Brown D, Anthony D, Lalueza-Fox C, Haak W, Pinhasi R, Reich D. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499-503. [Google Scholar] [CrossRef] |
| 25. | Fu Q, Posth C, Hajdinjak M, Petr M, Mallick S, Fernandes D, Furtwängler A, Haak W, Meyer M, Mittnik A, Nickel B, Peltzer A, Rohland N, Slon V, Talamo S, Lazaridis I, Lipson M, Mathieson I, Schiffels S, Skoglund P, Derevianko AP, Drozdov N, Slavinsky V, Tsybankov A, Cremonesi RG, Mallegni F, Gély B, Vacca E, Morales MRG, Straus LG, Neugebauer-Maresch C, Teschler-Nicola M, Constantin S, Moldovan OT, Benazzi S, Peresani M, Coppola D, Lari M, Ricci S, Ronchitelli A, Valentin F, Thevenet C, Wehrberger K, Grigorescu D, Rougier H, Crevecoeur I, Flas D, Semal P, Mannino MA, Cupillard C, Bocherens H, Conard NJ, Harvati K, Moiseyev V, Drucker DG, Svoboda J, Richards MP, Caramelli D, Pinhasi R, Kelso J, Patterson N, Krause J, Pääbo S, Reich D. The genetic history of ice age Europe. Nature. 2016;534:200-205. [Google Scholar] [CrossRef] |
| 26. | Schubert M, Lindgreen S, Orlando L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res Notes. 2016;9:1-7. [Google Scholar] [CrossRef] |
| 27. | Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754-1760. [Google Scholar] [CrossRef] |
| 28. | Peltzer A, Jäger G, Herbig A, Seitz A, Kniep C, Krause J, Nieselt K. EAGER: efficient ancient genome reconstruction. Genome Biol. 2016;17:1-14. [Google Scholar] [CrossRef] |
| 29. | Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078-2079. [Google Scholar] [CrossRef] |
| 30. | Jónsson H, Ginolhac A, Schubert M, Johnson PL, Orlando L. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics. 2013;29:1682-1684. [Google Scholar] [CrossRef] |
| 31. | Jun G, Wing MK, Abecasis GR, Kang HM. An efficient and scalable analysis framework for variant extraction and refinement from population-scale DNA sequence data. Genome Res. 2015;25:918-925. [Google Scholar] [CrossRef] |
| 32. | Kennett DJ, Plog S, George RJ, Culleton BJ, Watson AS, Skoglund P, Rohland N, Mallick S, Stewardson K, Kistler L, LeBlanc SA, Whiteley PM, Reich D, Perry GH. Archaeogenomic evidence reveals prehistoric matrilineal dynasty. Nat Commun. 2017;8:14115. [Google Scholar] [CrossRef] |
| 33. | Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, Zhao M, Chennagiri N, Nordenfelt S, Tandon A, Skoglund P, Lazaridis I, Sankararaman S, Fu Q, Rohland N, Renaud G, Erlich Y, Willems T, Gallo C, Spence JP, Song YS, Poletti G, Balloux F, van Driem G, de Knijff P, Romero IG, Jha AR, Behar DM, Bravi CM, Capelli C, Hervig T, Moreno-Estrada A, Posukh OL, Balanovska E, Balanovsky O, Karachanak-Yankova S, Sahakyan H, Toncheva D, Yepiskoposyan L, Tyler-Smith C, Xue Y, Abdullah MS, Ruiz-Linares A, Beall CM, Di Rienzo A, Jeong C, Starikovskaya EB, Metspalu E, Parik J, Villems R, Henn BM, Hodoglugil U, Mahley R, Sajantila A, Stamatoyannopoulos G, Wee JTS, Khusainova R, Khusnutdinova E, Litvinov S, Ayodo G, Comas D, Hammer MF, Kivisild T, Klitz W, Winkler CA, Labuda D, Bamshad M, Jorde LB, Tishkoff SA, Watkins WS, Metspalu M, Dryomov S, Sukernik R, Singh L, Thangaraj K, Pääbo S, Kelso J, Patterson N, Reich D. The Simons genome diversity project: 300 genomes from 142 diverse populations. Nature. 2016;538:201-206. [Google Scholar] [CrossRef] |
| 34. | Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, Reich D. Ancient admixture in human history. Genetics. 2012;192:1065-1093. [Google Scholar] [CrossRef] |
| 35. | Lazaridis I, Nadel D, Rollefson G, Merrett DC, Rohland N, Mallick S, Fernandes D, Novak M, Gamarra B, Sirak K, Connell S, Stewardson K, Harney E, Fu Q, Gonzalez-Fortes G, Jones ER, Roodenberg SA, Lengyel G, Bocquentin F, Gasparian B, Monge JM, Gregg M, Eshed V, Mizrahi A-S, Meiklejohn C, Gerritsen F, Bejenaru L, Blüher M, Campbell A, Cavalleri G, Comas D, Froguel P, Gilbert E, Kerr SM, Kovacs P, Krause J, McGettigan D, Merrigan M, Merriwether DA, O'Reilly S, Richards MB, Semino O, Shamoon-Pour M, Stefanescu G, Stumvoll M, Tönjes A, Torroni A, Wilson JF, Yengo L, Hovhannisyan NA, Patterson N, Pinhasi R, Reich D. Genomic insights into the origin of farming in the ancient Near East. Nature. 2016;536:419-424. [Google Scholar] [CrossRef] |
| 36. | Flegontov P, Altinişik NE, Changmai P, Rohland N, Mallick S, Adamski N, Bolnick DA, Broomandkhoshbacht N, Candilio F, Culleton BJ, Flegontova O, Friesen TM, Jeong C, Harper TK, Keating D, Kennett DJ, Kim AM, Lamnidis TC, Lawson AM, Olalde I, Oppenheimer J, Potter BA, Raff J, Sattler RA, Skoglund P, Stewardson K, Vajda EJ, Vasilyev S, Veselovskaya E, Hayes MG, O’Rourke DH, Krause J, Pinhasi R, Reich D, Schiffels S. Palaeo-Eskimo genetic ancestry and the peopling of Chukotka and North America. Nature. 2019;570:236-240. [Google Scholar] [CrossRef] |
| 37. | Jeong C, Balanovsky O, Lukianova E, Kahbatkyzy N, Flegontov P, Zaporozhchenko V, Immel A, Wang C-C, Ixan O, Khussainova E, Bekmanov B, Zaibert V, Lavryashina M, Pocheshkhova E, Yusupov Y, Agdzhoyan A, Koshel S, Bukin A, Nymadawa P, Turdikulova S, Dalimova D, Churnosov M, Skhalyakho R, Daragan D, Bogunov Y, Bogunova A, Shtrunov A, Dubova N, Zhabagin M, Yepiskoposyan L, Churakov V, Pislegin N, Damba L, Saroyants L, Dibirova K, Atramentova L, Utevska O, Idrisov E, Kamenshchikova E, Evseeva I, Metspalu M, Outram AK, Robbeets M, Djansugurova L, Balanovska E, Schiffels S, Haak W, Reich D, Krause J. The genetic history of admixture across inner Eurasia. Nat Ecol Evol. 2019;3:966-976. [Google Scholar] [CrossRef] |
| 38. | Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. [Google Scholar] [CrossRef] |
| 39. | Ringbauer H, Novembre J, Steinrücken M. Parental relatedness through time revealed by runs of homozygosity in ancient DNA. Nat Commun. 2021;12:5425. [Google Scholar] [CrossRef] |
| 40. | 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68-74. [Google Scholar] [CrossRef] |
| 41. | Bausch I. Prehistoric networks across the Korea Strait (5000–1000 BCE): ‘early globalization’ during the Jomon period in northwest Kyushu? In The Routledge Handbook of Archaeology and Globalization. London: Routledge; 2016. p.437-461. |
| 42. | Hudson MJ, Bausch IR, Robbeets M, Li T, White JA, Gilaizeau L. Bronze Age globalisation and Eurasian impacts on later Jōmon social change. J World Prehistory. 2021;34:121-158. [Google Scholar] [CrossRef] |
| 43. | Takahashi Y, Nasu H, Nakayama S, Tomooka N. Domestication of azuki bean and soybean in Japan: From the insight of archeological and molecular evidence. Breed Sci. 2023;73:117-131. [Google Scholar] [CrossRef] |

![]()
Copyright © 2026 Pivot Science Publications Corp. - unless otherwise stated | Terms and Conditions | Privacy Policy




