Abstract
Africa is the continent of our species’ origin and the deep history of humans is represented by African genetic variation. Through genetic studies, it has become evident that deep African population history is captured by relationships among African hunter-gatherers and that the world’s deepest population divergences occur among these groups. In this review, we look back at a study published by Cavalli-Sforza and colleagues in 1969 entitled “Studies on African Pygmies. I. A pilot investigation of Babinga Pygmies in the Central African Republic (with an analysis of genetic distances)”. The study analyzed 19 classical polymorphisms and found that the deepest divergences in African populations were represented by hunter-gatherer groups such as the southern African San and the central African rainforest hunter-gatherers. We repeated the original analyses from Cavalli-Sforza et al. [1] with about 22 thousand times more genome-wide genetic markers in populations similar to those included in the original study. Our high-resolution analyses gave similar results regarding the relationships of early-diverging African populations compared to the classical polymorphism analyses. This finding, however, does not imply that research has stagnated and that developments in technology and genetic methods over the last fifty years delivered no additional information regarding African history and adaptation. We review how technology and population genetic methods have advanced to give more detailed inferences about population structure, migrations, admixture patterns, timing of admixture, sex-biased admixture, and inferences of selection and adaptive introgression in rainforest hunter-gatherers and other African populations. We also comment on how sequencing of ancient DNA has influenced findings and deliberate on the progress and development of more complex models of African history, including alternatives to tree-models and the inference of possible archaic admixture in African populations. We review the growing complexity of our picture of population history in central Africa and Africa as a whole, emerging from genomic studies and other disciplines investigating human population history and adaptation. While data and knowledge are accumulating, certain populations and areas remain underrepresented in genomic research. Their inclusion, possibly also through ancient DNA studies, together with new methods of analysis and the testing of representative models of deep population history in Africa, will help to build a more complete picture of past population history in Africa.
Based on “Cavalli-Sforza LL, Zonta LA, Nuzzo F, Bernini L, de Jong WW, Meera Khan P, Ray AK, Went LN, Siniscalco M, Nijenhuis LE, van Loghem E, Modiano G. Studies on African Pygmies. I. A pilot investigation of Babinga Pygmies in the Central African Republic (with an analysis of genetic distances). Am J Hum Genet. 1969 May;21(3):252-274”.
Keywords
human evolution, admixture, population genetics, deep population history, Africa, rainforest hunter-gatherers, blood group systems
Introduction
Genetic studies of central African populations have substantially contributed to our understanding of human evolutionary history. A landmark study by Cavalli-Sforza and colleagues investigated patterns of genetic diversity among diverse African populations [1]. Their study focused on a west-central African population belonging to a group of populations, which at that time were called Pygmies. The word ‘Pygmy’, which derives from an ancient Greek word and was generally used to refer to individuals of small size, has been used as the name of human populations of small stature from the Congo Basin since the 1870s [2]. However, its use has been questioned because, among other reasons, it is a name given by foreigners to marginalised populations and not the name the people prefer to call themselves.
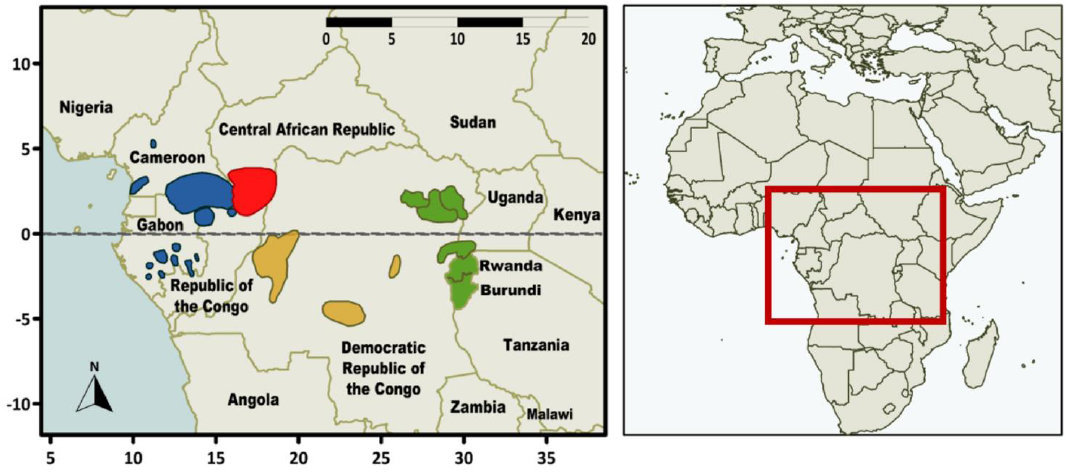
In the introduction to “African Pygmies” [3], Cavalli-Sforza draws a parallel between the use of the term ‘Pygmy’ and the term ‘Bushmen’; alternatives had been proposed to the latter name. Similarly, Cavalli-Sforza suggested some alternatives to the term, Pygmy, though none is deemed entirely satisfying. Almost forty years later, Hewlett addressed the same issue and presented the alternatives that are most commonly used today [4], such as ‘rainforest hunter-gatherers’ (abbreviated as RHGs), which we chose to apply in this review. However, using a different term does not remove the fact that RHGs are marginalised populations, and thus any term has to be used with care and respect. The naming of specific groups within larger populations is also a delicate matter. For example, the name ‘Babinga’ that was originally used by Cavalli-Sforza and colleagues [1] for a specific RHG group, is not used anymore, as it is a name given to them by neighbouring farmers and is perceived as being derogatory by this RHG group [3]. Instead, this RHG population from the Central African Republic (CAR) is referred to as ‘Aka’ or ‘Biaka’. Thus, the group name that we use in this review is Aka (Figure 1). Other terms used by Cavalli-Sforza et al. [1] have since become obsolete, such as ‘forest Negros’ or ‘Bushmen’ (replaced by San or a specific population name), and ‘Bantu’ or ‘Congo’ (which should be clarified as ‘Bantu-speaking populations’).
A common thread in research focusing on RHGs is the difficulty in finding appropriate criteria to categorize RHGs as a group [3,4]. RHG groups are not phenotypically or culturally homogeneous and they have highly diverse lifeways, languages, cultures and social interactions with neighbours [5,6]. Instead of a single criterion, lists of criteria have been proposed (see examples in [3] and [7]). One criterion that has proven unsatisfactory for distinguishing RHGs from other African populations is short stature, such as the threshold of 153 cm for males and 144 cm for females proposed by Cavalli-Sforza et al. [1]. Recent studies of diverse RHG groups have shown that RHG populations fall within the modern human height distribution, that variation in height across RHG populations is notably large, and that some non-RHG populations are as short as some RHG populations [8]. Language cannot be used as a classification either, since RHGs speak various languages, sometimes belonging to distinct linguistic families [5,6]. There is no unique shared language only spoken by RHG groups; instead RHG populations speak languages adopted from non-RHG populations, often Bantu-speaking farmers and often their neighbours [8].
The complex relationships and interactions between RHG populations and their neighbours have been well described in the literature and are a characteristic shared by all RHG populations. These relationships include the exchange of goods and labour, spiritual practices, gene flow, and in some cases a shared language [4]. Because of the complex relationships between the RHGs and their neighbours, several genetic studies include pairs of populations, in particular to investigate patterns of gene flow (for example [9–11]). This also means that population genetic studies of RHGs and their neighbours greatly benefit from cross-disciplinary collaborations with ethnologists, anthropologists, linguists and other researchers who have a good knowledge of the cultural background of the populations of interest.
Present-day African RHGs can be divided into three major groups: western, central, and eastern RHGs (abbreviated as wRHG, cRHG, and eRHG) (Figure 1), which are distinguished based on geography. The eastern and western groups have been shown to have a common genetic origin [12,13]. The western group, inhabiting Cameroon, the CAR, Gabon and the Republic of the Congo, includes populations such as the Baka, the Aka and the Bongo. The central group in the western and central parts of the Democratic Republic of Congo (DRC) includes populations such as the Twa and Cwa. The eastern group, living northeast of the DRC, and in Uganda and Rwanda, includes populations such as the Mbuti from the Ituri forest and the Twa from Uganda [8]. Some populations have been studied more than others. In particular, there is a bias towards studying ‘forest-oriented’ populations in the ethnographic literature [5]. Due to the political instability of the region, little is known about populations from the DRC [14]. In genetic studies, the Aka from the CAR and the Mbuti from eastern DRC, were sampled in the 1980s by Hewlett and Cavalli-Sforza and later included in the Human Genome Diversity Project (HGDP) [15]. They are commonly used as representatives of the western and eastern RHG populations, respectively. A large number of genetic studies have been done on RHG groups and some include a good representation of various RHG groups (for example [9,12,16–18]). An updated overview of genomic datasets from RHGs is included in Table 1. For a corresponding list prior to 2014, see Table 1 in [7].
Blood Group and Immunogenetic Markers Used in Genetic Studies
Blood group antigens are polymorphic traits inherited among individuals and populations. The existence of molecular genetic variations among human populations was first demonstrated in 1919 by characterizing the ABO blood group [19], which was the first human gene to be described. The subsequent identification of other blood group systems expanded the range of polymorphic markers. Since the 1950s, the analysis of these classical polymorphisms, in populations from broad geographic ranges, has been useful to investigate the origins and history of human populations worldwide. In total, 34 blood group systems have been recognized [20], which may reflect polymorphisms of red cell glycoproteins or carbohydrate epitopes on glycoproteins and glycolipids (e.g. ABO and Lewis systems), proteins (e.g. Diego, MNSs, and Duffy), and junctional complexes (e.g. Diego and MNSs).
Human immunoglobulin G (IgG) proteins are known for the large diversity of the variable domains of their heavy (gamma-globulin) chains, known as GM allotypes (the term allotype refers to any genetic variant of a protein). This highly polymorphic system, determined serologically by hemagglutination inhibition assays, is unique in its ability to characterize individuals and populations by specific sets of allotypes inherited in fixed combinations, or GM haplotypes [21]. Prior to the development of DNA sequencing techniques, GM haplotypes were used in bone marrow transplants, forensic medicine, and even paternity testing. Over four decades, GM haplotypes have been one of the most powerful tools in population genetics [22–24] and crucial to our understanding of the immunogenicity of antigenic determinants [21]. Large-scale screenings of human populations worldwide have uncovered considerable variability both in the kinds of GM haplotypes and in their frequencies. This led to new insights regarding genetic admixture, biogeography, ethno-anthropology, evolutionary biology and population genetics [22–26].
The frequencies of classical polymorphisms are useful tools for reconstructing the history of human migration, as they show different distributions between populations from different continents and within sub-continental populations. Although the analysis of classical markers does not allow estimation of the precise molecular diversity of human populations, knowledge of the gene frequencies of hundreds of populations worldwide can provide highly informative data for exploring spatial patterns of genetic diversity. The genetic patterns among human groups were summarised and described by Cavalli-Sforza et al. [22] in a landmark book demonstrating the worldwide geographic distribution of human genes and providing insights into human history. This book includes comprehensive tables and maps of the genetic information that the authors accumulated over fifty years; they examined more than 110 different polymorphisms such as blood groups, human leukocyte antigen (HLA) factors, immunoglobulin proteins, and DNA markers in over 1800 populations. Their work demonstrates the correlation between geography and particular genetic markers for detecting ancient migrations, for example the Bantu expansion in sub-Saharan Africa.
Case study: “Studies on African Pygmies. I. A pilot investigation of Babinga Pygmies in the Central African Republic (with an analysis of genetic distances)”
The study presented by Cavalli-Sforza et al. [1] included serological analyses of 175 Aka individuals from the CAR. To investigate their patterns of genetic diversity, haemoglobin variants, red cell enzymes (e.g. G6PD and haemoglobin S), blood group systems (e.g. ABO, Duffy, MNS, Rh, Kidd, P, Jk, Lutheran, Diego, Kell, Lewis, Hp, and Tf), and immunoglobulin allotypes (e.g. GM and Inv) were typed. Frequencies of certain blood group variants were similar to those among west-central African populations, such as for the Duffy null-allele or Fy(a-b-) with an estimated frequency of 100% in 163 tested individuals. The Duffy system was the first human blood group assigned to a specific autosomal chromosome [27]. The common Duffy alleles present striking patterns of geographic differentiation in human populations [28], as highlighted by Cavalli-Sforza et al. [22]. Individuals homozygous for the null variant, Fy(a-b-), have the Duffy-negative phenotype; absence of the Duffy protein on the surface of erythrocytes can confer protection against infection with Plasmodium vivax, one of the primary causative agents of malaria. Therefore, this study also provided important public health insights for African populations living in regions where malaria is endemic. The underlying mechanism of protection in Duffy-negative individuals is still unclear, however. Several studies have suggested through serological, molecular, and microscopic analyses that Duffy negativity is not completely protective against P. vivax infection and low levels of parasitemia can be detected in large surveys [29,30]. To better explain this low parasitemia and decipher P. vivax invasion mechanisms, future studies should investigate other red blood cell receptors of Duffy-negative individuals from west-central African populations.
The estimated frequencies of 29 classical polymorphisms typed in the Aka population were compared with frequencies in other African and worldwide populations obtained from the literature at the time [1]. The study showed that the Aka had typical ‘African’ genotypes. The frequencies were also used to calculate genetic distances between populations, which showed a bimodal African/non-African distribution. This confirmed results from previous studies, indicating that there were no increased genetic similarities between RHGs and other populations of short mean stature, such as the Southeast Asian Negrito groups. In their article Cavalli-Sforza et al. quote a sentence from Buettner-Janusch, 1966: “blood group gene frequencies suggest that Papuan Pygmies are simply short Papuans and African Pygmies are short Africans”.
Within African groups, the smallest genetic distances were found between agriculturist groups from the Congo and agriculturist groups living in the forest (Table 2A). These two groups were subsequently grouped as ‘central Africans’ in their analyses. A three-dimensional representation of genetic distances in figure 3 of Cavalli-Sforza et al. [1] indicated that the four main African groups, wRHG, eRHG, Khoe-San, and agriculturist central Africans, could be represented along three roughly equidistant axes. However, the distance of the eRHG and wRHG was smaller than the distance of Khoe-San to either group, supporting a common origin of RHG groups (Table 2A). The study also investigated group relationships with maximum-likelihood analyses of different tree topologies, and here again their ‘best tree’ supported a common origin of wRHG and eRHG populations. Aside from the closer association of wRHGs and eRHGs, an increased affinity between wRHGs and agriculturists was noted. The study went into further detail on the possibility of agriculturist admixture into the wRHGs and the effect that this admixture would have on genetic distances and topologies. They conducted further in-depth analysis on this, estimating possible mixture proportions by using the eRHGs and agriculturalist Central Africans as two parental populations. They estimated 45% gene flow from RHG agriculturist neighbours into the wRHG population. Although they strongly believed that their results indicated admixture between wRHGs and their agriculturist neighbours, they conceded that their dataset was too limited to make more definitive inferences.
To compare the results of analysing 29 classical polymorphisms presented in Cavalli-Sforza et al. [1] with current datasets, we examined over 640,000 genome-wide single-nucleotide polymorphisms (SNPs) in a similar set of African populations. Analogous to the 1969 study [1], we calculated Cavalli-Sforza’s chord genetic distance [31] between comparable pairs of African populations (Table 2B). We obtained smaller genetic distances than the ones reported in 1969 (Table 2A), perhaps due to the differences in genetic markers, both in number and type (here we used 411,903, predominantly non-genic, biallelic autosomal SNPs). Similar to the original study, the smallest genetic distances (of the four main groups) were found between wRHGs and agriculturalists and distances between eastern and western RHGs are smaller than distances to Khoe-San. It is important to note the role of natural selection in classical marker polymorphisms, which would have influenced the genetic distances reported in 1969 (Table 2A). For instance, sickle cell anaemia, thalassaemia, and certain enzyme polymorphisms are strongly associated with resistance to malaria in sub-Saharan African populations [32]. Likewise, the Duffy null-allele has been shown to protect against P. vivax infection and is at near fixation in sub-Saharan African populations like the Aka [1], while Fy(b+) and Fy(a+) are common in European and Asian populations, respectively [33]. In contrast, most of the genetic variations represented in genome-wide SNP arrays are in non-coding parts of the genome and are neutral or under background selection.
To visually compare our results with the three-dimensional representation in figure 3 of Cavalli-Sforza et al. [1], we plotted the calculated Cavalli-Sforza’s chord genetic distances (Table 2B) using the multidimensional scaling (MDS) algorithm (Figure 2A). For the genome-wide SNP dataset including the same populations, we also estimated the pairwise allele-sharing dissimilarity (ASD) matrix, a measure of genetic distance that is more commonly used today, between all pairs of individuals in the dataset and plotted the matrix using the MDS algorithm (Figure 2B) and a maximum likelihood tree using the TreeMix algorithm [34] (Figure 2C). Similar to Cavalli-Sforza et al. [1], the projections of both MDS plots show a strong genetic differentiation between wRHGs, eRHGs, Khoe-San, and western African agriculturalist populations (Figure 2), with affinity between wRHGs and western African agriculturalists that suggests admixture. Comparison of the 1969 results with today’s confirms our current knowledge on population structure and admixture between these African populations [9,16].
Analysis of Tree Topology in Africa
In 1969, Cavalli-Sforza and colleagues wrote about their tentative topology of the tree of modern humans that “the accumulation of further data, both on other markers and on related populations, should improve the resolution [of the phylogeny] to a satisfactory level”. While we have access to much more genetic data (see Table 1 for sequencing data only), the relationship between human lineages remains a much-discussed research topic [15,35]. It is now acknowledged that significant population structure already existed within Africa before the expansion towards the rest of the world, with extensive gene flow between African populations [36,37]. While recent gene flow during the past couple of thousand years is easier to characterize genetically and can be correlated with evidence from archaeology or linguistics, the study of deeper time periods is more complex. The utility of model testing and the importance of representative demographic models have been proposed and discussed [38] but applications to real data remain limited.
Today, the majority of approaches approximate African demographic history with trees that sometimes incorporate the possibility of gene flow between branches [15,35,38–42]. In a bifurcating tree representing modern human ancestry, the first divergence is generally found to be between the ancestors of the Khoekhoe and San populations living today in southern Africa (known as Khoe-San), and the ancestors of the rest of modern humans. The next divergence is between the ancestors of RHGs and the ancestors of the rest of modern humans. Further events include the divergence of the ancestors of western and eastern Africans, and finally of the branch ancestral to non-Africans. What has been demonstrated since Cavalli-Sforza et al. [1] is the common origin of a large number of wRHG populations [9], as well as the common origin of eastern and western RHGs [12,13,43]. West of the Congo Basin, a rapid diversification of RHG populations occurred in the past few thousand years, possibly correlated with the expansion of Bantu-speaking populations that fragmented territories [9,44].
The timing and modalities of these events have also been investigated using a variety of methods, genetic markers, and worldwide populations [15,35,37,45]. While the period 200-300 thousand years ago (kya) corresponds to the diversification of the ancestries found in present-day populations, it is difficult, if not impossible, to generate point estimates of the different events [35,46]. As an example, estimates for the divergence of the RHG branch vary from 350 to 70 kya [9,35], and the divergence of western and eastern RHGs was estimated at 20-80 kya [12,15,47–51].
Complex Patterns of Shared Ancestry Between African Hunter-gatherer Groups
Gene flow in deep time periods between geographically distant present day hunter-gatherer groups has been suggested by a number of studies [17,41,42]. Several lines of evidence point to pre-farming gene flow between eastern African hunter-gatherers and southern African Khoe-San [52,53]. This claim has been further substantiated with ancient DNA (aDNA) results from hunter-gatherers that lived in the region of present-day Malawi [42]. These hunter-gatherers had a genetic profile with intermediate levels from an “eastern African” genetic component associated with current-day eastern African hunter-gatherers from Tanzania and aDNA from East Africa, and a “southern African” genetic component, associated with southern African hunter-gatherers, both modern-day Khoe-San and ancient southern African Stone Age hunter-gatherers. The intermediate genetic profile of the Malawi hunter-gatherers suggests gene flow over deep time periods between eastern and southern African hunter-gatherers. This gene flow possibly represents an isolation-by-distance pattern that existed in the past, which connected hunter-gatherer groups across sub-Saharan Africa with low levels of step-wise gene flow between neighbouring groups, which gave rise to correlations between genetic and geographic distances between groups [54,55]. A similar pattern could therefore be extrapolated to include central African RHG groups. A recent whole-genome study of modern-day San groups showed higher genetic contributions from RHGs in San groups from the northern part of southern Africa (southern Angola) compared to more southerly San groups [45]. Indeed, when only ancient and modern-day hunter-gatherers are included in two-dimensional principal component analysis (PCA), there is a high correlation with geography (77%) which further points toward the possibility that hunter-gatherer groups were connected in the past through low levels of gene flow in and patterns of isolation-by-distance [54,55].
Shared ancestry and/or gene flow between RHG and other hunter-gatherer populations needs to be explored further in future research. Genetic information from ancient or modern representatives of populations that lived in the geographical areas between central African RHG populations and southern African San hunter-gatherer groups could contribute much to this debate. The majority of the populations in these areas today are Bantu-speaking agriculturists and most of the original inhabitants of these areas have been replaced or absorbed by the farming groups. The preservation of human remains and aDNA in west-central Africa is poor due to the acidic soil and tropical climate, thus remains from these parts are scarce and valuable. There are however a few marginal groups living in the DRC and Zambia, referred to as Batwa, Twa, or Cwa (see Figure 1 here and figure 2.1 in [1]). These groups are acknowledged by their farmer neighbours to be the original inhabitants of the region; some are hunter-gatherers, others are fishermen. Consequently, there is a possibility that they might still harbour a genetic component from the local pre-agricultural populations. Analysing their genetic composition with respect to Khoe-San and RHG contributions might offer valuable information to resolve long-standing questions regarding hunter-gatherer connections in the central and western parts of the continent.
Recent Patterns of Admixture in Central Africa
Today, descendants of the early-diverging human populations, the southern African Khoe-San, eastern and western RHGs, and eastern African hunter-gatherers such as the Hadza, are populations of small census sizes living in scattered regions. These populations all have some degree of admixture with groups of different genetic backgrounds, for example with Bantu-speaking agriculturalists, eastern African pastoralists or Eurasian groups. In general, admixture in RHG groups is mostly with their agriculturist neighbours; the amount of admixture varies across groups and is generally lower in eastern than in western groups. The mean admixture fraction is below 6% in Mbuti (eRHG) and Biaka (wRHG), and reaches almost 50% in Bezan (wRHG), while populations such as the Bakola and the Bongo have intermediate values [16]. Admixture fractions in RHGs tend to correlate with stature and social interaction with agricultural communities [9]. In contrast, admixture fractions from RHG groups into farming neighbours are generally low (ranging from 0.7 to 15.7% with a mean of 10.2%) [16].
Several investigations focused on the question of when these admixtures occurred. Archaeological records suggest hunter-gatherer occupation of the central African rainforests dating to 40 kya [56] and the first farmers only arrived in the region 3-5 kya [57]. Archaeological and linguistic studies indicate immediate interaction and trade between the RHGs and incoming farmers [3,58]. These early and close interactions were supported by biological studies dating the transfer of Helicobacter pylori bacterial strains from Bantu-speaking farmers to RHGs to about 4 kya [59]. Interestingly, genetic studies that estimated the timing of admixture events between RHGs and Bantu-speaking agriculturists found recent admixture dates using admixture-LD decay methods [16]. The estimated dates were more recent than 800 ya in all RHG populations (with a mean of 437 ya). These dates are much more recent than the initial contacts indicated by the archaeological record (3-5 kya). However, results showed that a single pulse model was not a good fit for the data and that continuous admixture or multiple pulses produced better fits. The recent dates could represent averages of several events, and could have been influenced by a large recent admixture pulse. Newer methods such as the MOSAIC software [60] that takes into account multiple pulses of admixture from multiple sources and the MetHis-ABC framework [61] for testing continuous contributions over time might further clarify and reconstruct the dynamics and timing of these events. A further possibility is that the original Bantu-speaker farmers, who expanded into the region, did indeed undergo admixture and genetic exchange with the local RHG, but that these initial Bantu-farming groups are not represented by current-day Bantu-speaking groups in the region. Several archaeological studies suggested a sharp decrease in farmer settlements across central Africa 1000-1600 BP, indicating a severe population crash [62,63]. Therefore, the initial Bantu-speaking groups that migrated into the region might have gone extinct or moved to other regions and were replaced by later waves of expansion and settlement in the region, leading to later expansion signals and admixture dates in the newly arrived groups [63]. Sequencing of aDNA of Bantu-speaking farmers from the region, before the population crash, might be able to shed more light on this question.
An aDNA study indeed confirmed an older interaction and genetic exchange between putative Bantu-speaking or West African agriculturist ancestors and RHG ancestors [64]. In the Shum Laka rock shelter in Cameroon, four individuals dated from about 8,000 to 3,000 ya were found to be genetically more similar to modern RHGs than to Bantu-speaking agriculturalists, who populate most of the region today. Although these individuals are not the direct ancestors of RHGs, they are clearly related to current-day wRHG populations, and they already showed admixture/affinity towards the West African genetic component harboured by current-day Bantu-speaking agriculturist groups [64]. This indicates population interactions between RHG and other West African groups during older time periods, preceding the introduction of agriculture.
Patterns of Sex-specific Admixture Within and Between Central African Populations
Ethnographers and population geneticists have long been interested in post-marital residence rules driving mobility after marriage, since these socio-cultural behaviours can shape the distribution of genetic variation within and among human populations [65]. The evolution of sociocultural behaviours determining post-marital residence rules and lineage transmission in African groups has been the subject of intense debate in cultural anthropology and ethnology [66]. The genetic effect of these socio-cultural behaviours is most directly reflected in the uniparental genetic markers, the mitochondrial genome (mtDNA) for the female lineages and the Y-chromosome for the male lineages [37]. In this context, population geneticists can determine how different sociocultural post-marital residence rules influenced the genetic variation observed today in mtDNA and Y-chromosome markers within existing populations, and estimate sex-biased admixture patterns.
Surprisingly, sex-specific genetic patterns in some sub-Saharan African populations apparently contradict population genetic expectations from post-marital residence rules [67]. Ethnographic studies showed that marriages usually occurred between RHG females and neighbouring males, but rarely or never between hunter-gatherer males and neighbouring females. One reason for this imbalance is that marrying a RHG female is more likely, due to lower bride compensations to be paid to the RHG families compared to agriculturalist families. The expected result would be RHG female gene flow into neighbouring populations. However, genetic evidence was compatible with a different pattern where male gene flow from neighbouring populations went into the RHG gene pool [68,69]. The two observations were reconciled in the following scenario: the neighbours’ social discrimination against the RHGs often results in RHG females marrying neighbouring males, but later divorcing and returning to their birth family with their children [69]. The children are then raised in the hunter-gatherer population, creating the observed sex-specific admixture pattern. Females might also move back to the hunter-gatherer population when their husbands from the neighbouring population died [69].
Among farming populations, genetic studies have evidenced high levels of genetic similarity in autosomal markers, and this genetic homogeneity is likely due to the recent demic expansion of Bantu-speaking populations concomitant with the expansion of agricultural techniques and Bantu languages starting about 4-5 kya [68-72]. However, if we focus on sex-specific genetic diversity patterns in these populations, genetic studies show more genetic differentiation among populations for genetic markers in the Y-chromosome than in the mtDNA [16,68-71]. These studies provide strong evidence for a higher female than male migration rate among farming populations from sub-Saharan Africa. This is consistent with the self-reported patrilocal practices in farming populations, where females move after marriage across communities and populations much more often than males [69,72]. Sex-specific genetic patterns in farming populations may also be linked to the practice of polygyny, which reduces the genetic diversity of male lineages compared to female lineages [65].
What More Can We Infer from Genomic Data and Current Methods?
Our repeat of the original analyses from Cavalli-Sforza et al. [1] with 22,372 times more genetic markers (Figure 2), gave very similar results regarding the relationships of early-diverging African populations. However, this does not imply that research has stagnated and that developments in technologies and methods over the past fifty years delivered no additional information regarding RHG history and adaptation, and African population history as a whole. Technologies that developed denser marker typing such as short tandem repeat (STR) analyses and SNP arrays made whole-genome representation available by effectively tagging genome representative haplotypes. Aside from denser genome-wide coverage, these technologies also gave us access to neutral variation in the genome, which better represent population demographic history compared to protein markers, which are subject to selection. Genome-wide autosomal research started with STR markers, which immediately highlighted the unique position and divergence of African hunter-gatherer populations with regards to other African populations [9,17,73]. The use of newly developed, cluster-based methods has delivered fine-scale inferences on population structure and possible admixture. Together with algorithmic methods, such as PCA, it formed the basis of many ensuing African population structure studies based on genome-wide genotype data [16,34,52,74,75]. Those studies all confirmed the unique position of hunter-gatherer groups with regard to population structure in Africa and also suggested various levels of admixture between different hunter-gatherer groups and their neighbours.
Although clustering and algorithmic methods can only uncover suggestions of admixture, new formal tests of admixture based on allele sharing in simple topologies allow formal testing of admixture [76]. Therefore, admixtures between RHG and other populations could be formally tested, and admixture fractions could be estimated [64]. Methods dating admixture events, based on LD-admixture patterns [76] or the lengths of local ancestry fragments [77,78] were used to estimate specific timings of major admixture events [9,12,16]. Inference of sex-biased admixture based on comparing X-chromosomal admixture ratios to autosomal ratios [79] also contributed to our knowledge about population histories. Sex-biased admixture patterns in RHG populations could be inferred using these and other methods such as approximate Bayesian computation (ABC) analyses [69] to complement inferences already obtained by uniparental markers [9,13,16,67–69]. Thus, although admixture with RHG neighbour groups was a possibility that could only be alluded to in Cavalli Sforza et al. [1], advances in the past fifty years, provided much more information regarding formal tests of admixture, admixture fractions, admixture timing and sex-biased admixture patterns.
Population size is another aspect of demography that has been extensively studied in the past decade, in particular using methods like multiple sequential Markovian coalescent (MSMC) that infer effective population size over time [80]. There is evidence that the effective population size of RHG populations like the Baka, BaTwa, Biaka and Mbuti was large for most of their past—similar to or larger than the size of neighbouring agriculturalist populations [11,15,35]. This contrasts with the small census sizes of RHG observed today, and suggests that RHG formed a metapopulation connected with gene flow. Another observation is that RHG population sizes have been decreasing over the past 10,000 years while agriculturalist populations have been increasing [11,12,15,35]; this predated the shift to agriculture in central Africa, and suggests that the ancestors of agriculturalists experienced an increase of population size prior to the adoption of agriculture [16].
Aside from demographic inferences, genome-wide representation also provides the opportunity of scanning genomes for regions that were subject to selection in specific populations. Since an unbiased estimate of genetic diversity is not obtained using array-based genetic markers that usually specifically target intermediate frequency polymorphisms in reference populations, the classical population genetic selection tests based on the site frequency spectrum (SFS) were not possible in array-based studies. However, much information regarding selective events and adaptations in populations have been acquired, mostly through two types of methods for genome-wide selection scans regularly applied to SNP-array data. These include methods that estimate frequency differences at specific genomic positions between populations [81] and methods for estimating haplotype homozygosity surrounding specific alleles [82,83]. Using these types of selection scans, interesting signals have been identified in hunter-gatherer populations in Africa [11,52,84]. In particular, numerous genetic studies tried to identify the genetic basis of height in RHGs and/or to test hypotheses positing an adaptive advantage of short stature [18,85–87]. Immune response genes were also found to be a recurring target of adaptation in RHGs. Moreover, Patin et al. [74] identified evidence of adaptive introgression in western African Bantu-speaking agriculturists, showing an excess of RHG ancestry in the HLA region. This indicated that central African agriculturalist populations might have acquired immunity-related alleles from RHG groups that they admixed with, which helped them to adapt to the different pathogen environments they encountered during their expansion into the RHG geographical areas.
Although genome-wide genotype panels are very useful for many kinds of analysis and yield valuable inferences, SNP arrays do not represent an unbiased sample of genomic diversity. Specific reference populations were used to select the genetic markers represented on SNP arrays. Genetic markers that are polymorphic in these reference populations may not be polymorphic in other populations. Similarly, loci that were monomorphic in the reference populations would not have been chosen to be represented in the SNP array, but may be polymorphic in other populations. This may generate allele frequency biases that could influence allele frequencies in different populations and lead to the identification of spurious demographic events and selection signatures. African populations, especially hunter-gatherers and other marginalized populations, were not well represented in reference panels used to select haplotype tagging markers of SNP arrays; therefore, their unbiased genetic diversity was hidden to a large extent. Recent full genome sequencing studies highlighted the extensive diversity in hunter-gatherer populations (Table 1) [45,47,88], and led to the finer estimation of deep structure and major population split-time inferences in Africa. Bifurcating tree models however remain an over-simplification of a more complex underlying history. Future studies should strive to use available full genome data to test more complex demographic scenarios [38–40,89,90] that allow the inference of pulses of admixture and/or continuous gene flow. These models should ideally not just include modern-day populations but also aDNA results as reference points or prior information. Complex demographic models are often inferred by composite likelihood methods [91–94]. More recent approaches have attempted to move beyond tree-like approaches (e.g. [95–97]). By combining simulations with machine learning algorithms, ABC approaches allow researchers to specify virtually any model and to formally choose between models [49,98,99]. Recent studies have started to apply these methods to entire genomes [88].
Ancient DNA studies have demonstrated that admixture had an influence on most present-day hunter-gatherer and neighbouring agriculturalist populations [41,64,100,101], and that our perceptions and inferences about simple major mode admixture timing does not always capture this complex history [64]. Ideally, models should strive to include possible events of archaic admixture. The possible contribution of unknown African archaic populations (deeply diverging past populations who did not leave distinct populations as descendants today) have been highlighted, proposed and inferred by several studies [36,41,42,64,102–105]. Despite the growing number of genomes from African populations and the new statistical tools developed for detecting archaic introgression, there are still obstacles to detecting signals of putative archaic introgression. These include the lack of an archaic African genome, the lack of reference genomes without any admixture, the lack of an appropriate ‘outgroup’ population particularly when the target is an early-diverging population, and sample sizes of high-coverage genomes that remain low for many African populations [106], which limits the power of allele frequency-based methods.
Besides the limitations, cumulative genetic evidence is suggesting a complex history of archaic admixture or deep structure within African populations. Signals of putative archaic admixture were also identified in some RHG populations, such as the Biaka, the Baka, and the Mbuti, using different methods, such as S* [87,107], which identifies long LD fragments, IMa3, which reconstructs the entire genealogy of several hunter-gatherer populations [108], and ABC [88]. While the modalities of archaic introgression/s remain to be clarified, a number of studies are compatible with independent events into the ancestors of several modern populations such as RHGs, Khoe-San and west Africans [88,102,108]. Several studies are compatible with a divergence of the estimated archaic source from the ancestors of modern humans around the time of the split with the ancestors of Neanderthals, about 600 kya years ago, though the divergence estimates range from 1.2 million years to 300 kya [9,35]. In future studies it will be important to clarify different scenarios of admixture by more systematic testing of RHG and other African populations for archaic admixture.
Future Directions and New Technologies
Other ‘omics’ studies have recently complemented genomic research to address questions about population history and adaptation. Epigenomics, transcriptomics and microbiome studies have highlighted that RHGs possess specific adaptations compared to their neighbours [109]. A landmark methylome study combined datasets of SNP-array data and DNA methylation profiles for RHG and neighbouring agriculturalist populations [110]. To contrast effects associated with the recent change of habitats versus historical lifestyle, they compared agriculturalists living in urban areas to those in the rainforest. They showed that both the current habitat and the historical lifestyle had an effect on the methylome, but they encompass different biological functions. Current habitats were found to have an influence on methylation profiles regarding immune and cellular function while the historical lifestyle pattern influenced developmental processes. Furthermore, they found that the historical lifestyle effect was more strongly correlated with changes in the DNA.
Several gut microbiome studies among wRHGs, Baka, Bagyeli and Aka, and neighbours appeared somewhat contradictory; however all studies point to higher parasitic loads in RHGs [111–113]. A transcriptomic study by Harrison et al. [10] tested the transcriptional response of peripheral blood mononuclear cells to bacterial and viral stimuli. They compared Batwa (eRHG) and Bakiga (agriculturalists) and found larger divergences between these groups in their response to viral stimuli and stronger positive selection signals in RHGs. These studies revealed unexpected findings for comparisons between hunter-gatherer vs. farmer groups, since a common theory is that the shift to agriculture increased the occurrence of diseases and parasitic infections [114] and therefore also pressure on the immune system. Future genomic and other ‘omic’ studies need to be done to provide more data for further inferences about lifestyles and adaptations of RHG communities and their neighbours.
Conclusions
Since the 1969 Cavalli-Sforza et al. study of the Aka [1], with a limited number of serogenetic markers and only a few comparative populations, central African populations have become better represented in genetic and genomic datasets. This allowed a more accurate description of their genetic diversity and progress in the reconstruction of their evolutionary history and relationships to other populations. In particular, different models of population topologies could be tested, complex patterns of sex-biased admixture have been identified, admixture times and fractions with different populations have been estimated, and selection signals and adaptive introgression have been identified. While data and knowledge are accumulating, certain populations and areas still remain underrepresented in genomic research. Their inclusion in future studies, hopefully including aDNA sequencing, will continue to help build a more complete picture of past population structure in Africa. As we learn more about human history, demographic models become more and more complex, incorporating for example the possibility of archaic admixture in Africa. The Cavalli-Sforza et al. study together with similar studies using classical genetic markers, focusing on the demographic history of RHGs and their role in deep African history, established a foundation for future population genetic studies that will remain a topic of interest for many years to come.
Declarations
Availability of Data and Material
For the genome-wide genotype data from the [74] study (EGA accessory numbers: EGAD00010001209), data access was granted via European GenomePhenome Archive (EGA) by the GEH Data Access Committee EGAC00001000139 (date of approval: 26 April 2021). Data from the [52] study is downloadable from: http://jakobssonlab.iob.uu.se/data for non-commercial research use.
Funding
C.F-L. and C.S. were funded by the European Research Council (ERC StG AfricanNeo, grant no. 759933 to C.S.). C.F-L. was funded by the Bertil Lundman Foundation for Anthropological Studies, the Royal Physiographic Society of Lund (Nilsson-Ehle Endowments), and the Sven and Lilly Lawski’s Foundation (postdoctoral scholarship). G.B. was funded by the Knut and Alice Wallenberg foundation (support to Mattias Jakobsson) and the Swedish Research Council (no: 2018-05537 to Mattias Jakobsson).
Competing Interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgement
We thank the editors for the invitation to participate in this Research Topic. We would like to thank Paul Verdu for enlightening discussions and the two reviewers for their insightful comments. The computations and data handling were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at UPPMAX (project number: SNIC 2019/8-348) partially funded by the Swedish Research Council through grant agreement no. 2018-05973.
Table 1 Available whole-genome and exome sequences from RHG populations (ancient and modern samples). List of publications updated up to April 2021. wRHG: western rainforest hunter-gatherers, eRHG: eastern rainforest hunter-gatherers.
| Population name |
Sample Size |
Country |
Type of Data (Mean Overage) |
Reference |
| Ancient DNA |
|
|
|
|
| Shum Laka |
4 |
Cameroon |
genome (0.7 to 7.7X) |
[64] |
| Modern DNA |
|
|
|
|
| “Congo Pygmies” |
3 |
“Congo” (DRC) |
genome |
[115] |
| Baka (wRHG) |
3 |
Cameroon |
genome (>60X) |
[104] |
| Baka (wRHG) |
2 |
Cameroon |
genome (~43X) |
[47] |
| Baka (wRHG) |
1 |
Gabon |
genome (32X) |
[88] |
| Baka (wRHG) |
20 |
Gabon |
genome (~6.5X) |
[11] |
| Bakola (wRHG) |
1 |
Cameroon |
genome (>60X) |
[104] |
| Bakola (wRHG) |
2 |
Cameroon |
genome (~43X) |
[47] |
| Bedzan (wRHG) |
1 |
Cameroon |
genome (>60X) |
[104] |
| Bedzan (wRHG) |
2 |
Cameroon |
genome (~43X) |
[47] |
| Biaka (wRHG) |
4 |
CAR |
genome (~60.5X) |
[103] |
| Biaka (wRHG) |
2 |
CAR |
genome (high-coverage) |
[48] |
| Biaka (wRHG) |
20 |
CAR |
genome (~35X) |
[15] |
| Mbuti (eRHG) |
1 |
DRC |
genome (~20X) |
[116] |
| Mbuti (eRHG) |
4 |
DRC |
genome (high-coverage) |
[48] |
| Mbuti (eRHG) |
8 |
DRC |
genome (~35X) |
[15] |
| Babongo (wRHG) |
21 |
Gabon (center) |
exome (~40X) |
[11] |
| Babongo (wRHG) |
27 |
Gabon (east) |
exome (~40X) |
[11] |
| Babongo (wRHG) |
33 |
Gabon (south) |
exome (~40X) |
[11] |
| Baka (wRHG) |
30 |
Gabon |
exome (~40X) |
[11] |
| Baka (wRHG) |
100 |
Cameroon |
exome (~68X) |
[50] |
| Bakoya (wRHG) |
26 |
Gabon |
exome (~40X) |
[11] |
| Bezan (wRHG) |
38 |
Cameroon |
exome (~40X) |
[11] |
| Batwa (eRHG) |
51 |
Uganda |
exome |
[50] |
References
| 1. |
Cavalli-Sforza LL, Zonta LA, Nuzzo F, Bernini L, de Jong WW, Meera Khan P, et al. Studies on African Pygmies. I. A pilot investigation of Babinga Pygmies in the Central African Republic (with an analysis of genetic distances). Am J Hum Genet. 1969;21:252-274.
[Google Scholar]
|
| 2. |
Bahuchet S. L’invention des Pygmées. Cahiers D’études Africaines. 1993;33:153-181.
[Google Scholar]
[CrossRef]
|
| 3. |
Cavalli-Sforza LL. African Pygmies. Massachusetts, USA: Academic Press; 1986.
[Google Scholar]
|
| 4. |
Hewlett BS. Hunter-Gatherers of the Congo Basin: Cultures, Histories, and Biology of African Pygmies. New York, USA: Routledge; 2014.
[Google Scholar]
|
| 5. |
Bahuchet S. Cultural diversity of African Pygmies. In: Hewlett BS, editor. Hunter-Gatherers of the Congo Basin: Cultures, Histories, and Biology of African Pygmies. New York, USA: Routledge; 2014. p.1-30.
[Google Scholar]
[CrossRef]
|
| 6. |
Bahuchet S. Changing language, remaining pygmy. Hum Biol. 2012;84(1):11-43.
[Google Scholar]
[CrossRef]
|
| 7. |
Verdu P. Population Genetics of Central African Pygmies and Non-Pygmies. In: Hewlett BS, editor. Hunter-Gatherers of the Congo Basin. Cultures, Histories, and Biology of African Pygmies. New York, USA: Routledge; 2014. p.31-59.
[Google Scholar]
[CrossRef]
|
| 8. |
Verdu P. African Pygmies. Curr Biol. 2016;26:R12-R14.
[Google Scholar]
[CrossRef]
|
| 9. |
Verdu P, Austerlitz F, Estoup A, Vitalis R, Georges M, Théry S, et al. Origins and genetic diversity of pygmy hunter-gatherers from Western Central Africa. Curr Biol. 2009;19:312-318.
[Google Scholar]
[CrossRef]
|
| 10. |
Harrison GF, Sanz J, Boulais J, Mina MJ, Grenier J-C, Leng Y, et al. Natural selection contributed to immunological differences between hunter-gatherers and agriculturalists. Nat Ecol Evol. 2019;3:1253-1264.
[Google Scholar]
[CrossRef]
|
| 11. |
Lopez M, Choin J, Sikora M, Siddle K, Harmant C, Costa HA, et al. Genomic evidence for local adaptation of hunter-gatherers to the African rainforest. Curr Biol. 2019;29:2926-2935.
[Google Scholar]
[CrossRef]
|
| 12. |
Patin E, Laval G, Barreiro LB, Salas A, Semino O, Santachiara-Benerecetti S, et al. Inferring the Demographic History of African Farmers and Pygmy Hunter–Gatherers Using a Multilocus Resequencing Data Set. PLoS Genet. 2009;5:e1000448.
[Google Scholar]
[CrossRef]
|
| 13. |
Batini C, Lopes J, Behar DM, Calafell F, Jorde LB, van der Veen L, et al. Insights into the demographic history of African Pygmies from complete mitochondrial genomes. Mol Biol Evol. 2011;28:1099-1110.
[Google Scholar]
[CrossRef]
|
| 14. |
Froment A. Human Biology and Health of African Rainforest Inhabitants. In: Hewlett BS, editor. Hunter-Gatherers of the Congo Basin. Cultures, Histories, and Biology of African Pygmies. New York, USA: Routledge; 2014. p.117-164.
[Google Scholar]
[CrossRef]
|
| 15. |
Bergström A, McCarthy SA, Hui R, Almarri MA, Ayub Q, Danecek P, et al. Insights into human genetic variation and population history from 929 diverse genomes. Science. 2020;367:eaay5012.
[Google Scholar]
[CrossRef]
|
| 16. |
Patin E, Siddle KJ, Laval G, Quach H, Harmant C, Becker N, et al. The impact of agricultural emergence on the genetic history of African rainforest hunter-gatherers and agriculturalists. Nat Commun. 2014;5:3163.
[Google Scholar]
[CrossRef]
|
| 17. |
Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, et al. The Genetic Structure and History of Africans and African Americans. Science. 2009;324:1035-1044.
[Google Scholar]
[CrossRef]
|
| 18. |
Perry GH, Foll M, Grenier J-C, Patin E, Nédélec Y, Pacis A, et al. Adaptive, convergent origins of the pygmy phenotype in African rainforest hunter-gatherers. Proc Natl Acad Sci USA. 2014;111:E3596-E3603.
[Google Scholar]
[CrossRef]
|
| 19. |
Hirszfeld L, Hirszfeld H. Essai d’application des méthodes sérologiques au problème des races. Anthropologie. 1919;29:505-537.
[Google Scholar]
|
| 20. |
Storry JR, Castilho L, Daniels G, Flegel WA, Garratty G, de Haas M, et al. International Society of Blood Transfusion Working Party on red cell immunogenetics and blood group terminology: Cancun report (2012). Vox Sang. 2014;107:90-96.
[Google Scholar]
[CrossRef]
|
| 21. |
Lefranc M-P, Lefranc G. Human Gm, Km, and Am allotypes and their molecular characterization: a remarkable demonstration of polymorphism. Methods Mol Biol. 2012;882:635-680.
[Google Scholar]
[CrossRef]
|
| 22. |
Cavalli-Sforza LL, Cavalli-Sforza L, Menozzi P, Piazza A. The History and Geography of Human Genes. New Jersey, USA: Princeton University Press; 1994.
[Google Scholar]
|
| 23. |
Dugoujon J-M, Hazout S, Loirat F, Mourrieras B, Crouau-Roy B, Sanchez-Mazas A. GM haplotype diversity of 82 populations over the world suggests a centrifugal model of human migrations. Am J Phys Anthropol. 2004;125:175-192.
[Google Scholar]
[CrossRef]
|
| 24. |
Steinberg AG, Cook CE. The distribution of the human immunoglobulin allotypes. USA: Oxford University Press; 1981.
[Google Scholar]
|
| 25. |
Fortes-Lima C, Dugoujon J-M, Hernández CL, Reales G, Calderón R. Immunoglobulin genes in Andalusia (Spain). Genetic diversity in the Mediterranean space. C R Biol. 2014;337:646-656.
[Google Scholar]
[CrossRef]
|
| 26. |
Sanchez-Mazas A, Fernandez-Viña M, Middleton D, Hollenbach JA, Buhler S, Di D, et al. Immunogenetics as a tool in anthropological studies. Immunology. 2011;133:143-164.
[Google Scholar]
[CrossRef]
|
| 27. |
Donahue RP, Bias WB, Renwick JH, McKusick VA. Probable assignment of the Duffy blood group locus to chromosome 1 in man. Proc Natl Acad Sci USA. 1968;61:949-955.
[Google Scholar]
[CrossRef]
|
| 28. |
Howes RE, Patil AP, Piel FB, Nyangiri OA, Kabaria CW, Gething PW, et al. The global distribution of the Duffy blood group. Nat Commun. 2011;2:266.
[Google Scholar]
[CrossRef]
|
| 29. |
Kano FS, de Souza AM, de Menezes Torres L, Costa MA, Souza-Silva FA, Sanchez BAM, et al. Susceptibility to Plasmodium vivax malaria associated with DARC (Duffy antigen) polymorphisms is influenced by the time of exposure to malaria. Sci Rep. 2018;8:13851.
[Google Scholar]
[CrossRef]
|
| 30. |
Popovici J, Roesch C, Rougeron V. The enigmatic mechanisms by which Plasmodium vivax infects Duffy-negative individuals. PLoS Pathog. 2020;16:e1008258.
[Google Scholar]
[CrossRef]
|
| 31. |
Cavalli-Sforza LL, Edwards AWF. Phylogenetic analysis: Models and estimation procedures. Evolution. 1967;21:550-570.
[Google Scholar]
[CrossRef]
|
| 32. |
Williams TN, Weatherall DJ. World distribution, population genetics, and health burden of the hemoglobinopathies. Cold Spring Harb Perspect Med. 2012;2:a011692.
[Google Scholar]
[CrossRef]
|
| 33. |
McManus KF, Taravella AM, Henn BM, Bustamante CD, Sikora M, Cornejo OE. Population genetic analysis of the DARC locus (Duffy) reveals adaptation from standing variation associated with malaria resistance in humans. PLoS Genet. 2017;13:e1006560.
[Google Scholar]
[CrossRef]
|
| 34. |
Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8:e1002967.
[Google Scholar]
[CrossRef]
|
| 35. |
Hollfelder N, Breton G, Sjödin P, Jakobsson M. The deep population history in Africa. Hum Mol Genet. 2021;30:R2-R10.
[Google Scholar]
[CrossRef]
|
| 36. |
Schlebusch CM, Jakobsson M. Tales of Human Migration, Admixture, and Selection in Africa. Annu Rev Genomics Hum Genet. 2018;19:405-428.
[Google Scholar]
[CrossRef]
|
| 37. |
Cavalli-Sforza LL, Feldman MW. The application of molecular genetic approaches to the study of human evolution. Nat Genet. 2003;33:266-275.
[Google Scholar]
[CrossRef]
|
| 38. |
Scerri EML, Chikhi L, Thomas MG. Beyond multiregional and simple out-of-Africa models of human evolution. Nat Ecol Evol. 2019;3:1370-1372.
[Google Scholar]
[CrossRef]
|
| 39. |
Henn BM, Steele TE, Weaver TD. Clarifying distinct models of modern human origins in Africa. Curr Opin Genet Dev. 2018;53:148-156.
[Google Scholar]
[CrossRef]
|
| 40. |
Scerri EML, Thomas MG, Manica A, Gunz P, Stock JT, Stringer C, et al. Did Our Species Evolve in Subdivided Populations across Africa, and Why Does It Matter? Trends Ecol Evol. 2018;33:582-594.
[Google Scholar]
[CrossRef]
|
| 41. |
Schlebusch CM, Malmström H, Günther T, Sjödin P, Coutinho A, Edlund H, et al. Southern African ancient genomes estimate modern human divergence to 350,000 to 260,000 years ago. Science. 2017;358:652-655.
[Google Scholar]
[CrossRef]
|
| 42. |
Skoglund P, Thompson JC, Prendergast ME, Mittnik A, Sirak K, Hajdinjak M, et al. Reconstructing Prehistoric African Population Structure. Cell. 2017;171:59-71.
[Google Scholar]
[CrossRef]
|
| 43. |
Destro-Bisol G, Coia V, Boschi I, Verginelli F, Cagliá A, Pascali V, et al. The analysis of variation of mtDNA hypervariable region 1 suggests that Eastern and Western Pygmies diverged before the Bantu expansion. Am Nat. 2004;163:212-226.
[Google Scholar]
[CrossRef]
|
| 44. |
Quintana-Murci L, Quach H, Harmant C, Luca F, Massonnet B, Patin E, et al. Maternal traces of deep common ancestry and asymmetric gene flow between Pygmy hunter-gatherers and Bantu-speaking farmers. Proc Natl Acad Sci USA. 2008;105:1596-1601.
[Google Scholar]
[CrossRef]
|
| 45. |
Schlebusch CM, Sjödin P, Breton G, Günther T, Naidoo T, Hollfelder N, et al. Khoe-San Genomes Reveal Unique Variation and Confirm the Deepest Population Divergence in Homo sapiens. Mol Biol Evol. 2020;37:2944-2954.
[Google Scholar]
[CrossRef]
|
| 46. |
Bergström A, Stringer C, Hajdinjak M, Scerri EML, Skoglund P. Origins of modern human ancestry. Nature. 2021;590:229-237.
[Google Scholar]
[CrossRef]
|
| 47. |
Fan S, Kelly DE, Beltrame MH, Hansen MEB, Mallick S, Ranciaro A, et al. African evolutionary history inferred from whole genome sequence data of 44 indigenous African populations. Genome Biol. 2019;20:82.
[Google Scholar]
[CrossRef]
|
| 48. |
Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature. 2016;538:201-206.
[Google Scholar]
[CrossRef]
|
| 49. |
Veeramah KR, Wegmann D, Woerner A, Mendez FL, Watkins JC, Destro-Bisol G, et al. An early divergence of KhoeSan ancestors from those of other modern humans is supported by an ABC-based analysis of autosomal resequencing data. Mol Biol Evol. 2012;29:617-630.
[Google Scholar]
[CrossRef]
|
| 50. |
Lopez M, Kousathanas A, Quach H, Harmant C, Mouguiama-Daouda P, Hombert J-M, et al. The demographic history and mutational load of African hunter-gatherers and farmers. Nat Ecol Evol. 2018;2:721-730.
[Google Scholar]
[CrossRef]
|
| 51. |
Breton G. Human demographic history: Insights on the human past based on genomes from Southern through Central Africa. PhD Thesis: Acta Universitatis Upsaliensis; 2020.
|
| 52. |
Schlebusch CM, Skoglund P, Sjödin P, Gattepaille LM, Hernandez D, Jay F, et al. Genomic variation in seven Khoe-San groups reveals adaptation and complex African history. Science. 2012;338:374-379.
[Google Scholar]
[CrossRef]
|
| 53. |
Naidoo T, Xu J, Vicente M, Malmström H, Soodyall H, Jakobsson M, et al. Y-Chromosome Variation in Southern African Khoe-San Populations Based on Whole-Genome Sequences. Genome Biol Evol. 2020;12:1031-1039.
[Google Scholar]
[CrossRef]
|
| 54. |
Vicente M, Schlebusch CM. African population history: an ancient DNA perspective. Curr Opin Genet Dev. 2020;62:8-15.
[Google Scholar]
[CrossRef]
|
| 55. |
Vicente M, Jakobsson M, Ebbesen P, Schlebusch CM. Genetic Affinities among Southern Africa Hunter-Gatherers and the Impact of Admixing Farmer and Herder Populations. Mol Biol Evol. 2019;36:1849-1861.
[Google Scholar]
[CrossRef]
|
| 56. |
Cornelissen E. Human Responses to Changing Environments in Central Africa Between 40,000 and 12,000 B.P. Journal of World Prehistory. 2002;16:197-235.
[Google Scholar]
[CrossRef]
|
| 57. |
Bostoen K. The Bantu Expansion. Oxford Research Encyclopedia of African History. Oxford, UK: Oxford University Press; 2018. p.1-28.
[Google Scholar]
[CrossRef]
|
| 58. |
Blench R. Archaeology, Language, and the African Past. Maryland, USA: Rowman Altamira; 2006.
[Google Scholar]
|
| 59. |
Nell S, Eibach D, Montano V, Maady A, Nkwescheu A, Siri J, et al. Recent acquisition of Helicobacter pylori by Baka pygmies. PLoS Genet. 2013;9:e1003775.
[Google Scholar]
[CrossRef]
|
| 60. |
Salter-Townshend M, Myers S. Fine-Scale Inference of Ancestry Segments Without Prior Knowledge of Admixing Groups. Genetics. 2019;212:869-889.
[Google Scholar]
[CrossRef]
|
| 61. |
Fortes-Lima CA, Laurent R, Thouzeau V, Toupance B, Verdu P. Complex genetic admixture histories reconstructed with Approximate Bayesian Computation. Mol Ecol Resour. 2021;21:1098-1117.
[Google Scholar]
[CrossRef]
|
| 62. |
Oslisly R, White L, Bentaleb I, Favier C, Fontugne M, Gillet J-F, et al. Climatic and cultural changes in the west Congo Basin forests over the past 5000 years. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120304.
[Google Scholar]
[CrossRef]
|
| 63. |
Seidensticker D, Hubau W, Verschuren D, Fortes-Lima C, de Maret P, Schlebusch CM, et al. Population collapse in Congo rainforest from 400 CE urges reassessment of the Bantu Expansion. Sci Adv. 2021;7.
[Google Scholar]
[CrossRef]
|
| 64. |
Lipson M, Ribot I, Mallick S, Rohland N, Olalde I, Adamski N, et al. Ancient West African foragers in the context of African population history. Nature. 2020;577:665-670.
[Google Scholar]
[CrossRef]
|
| 65. |
Heyer E, Chaix R, Pavard S, Austerlitz F. Sex-specific demographic behaviours that shape human genomic variation. Mol Ecol. 2012;21:597-612.
[Google Scholar]
[CrossRef]
|
| 66. |
MacDonald DH, Hewlett BS. Reproductive Interests and Forager Mobility. Curr Anthropol. 1999;40:501-524.
[Google Scholar]
[CrossRef]
|
| 67. |
Destro-Bisol G, Donati F, Coia V, Boschi I, Verginelli F, Caglià A, et al. Variation of female and male lineages in sub-Saharan populations: the importance of sociocultural factors. Mol Biol Evol. 2004;21:1673-1682.
[Google Scholar]
[CrossRef]
|
| 68. |
Berniell-Lee G, Calafell F, Bosch E, Heyer E, Sica L, Mouguiama-Daouda P, et al. Genetic and demographic implications of the Bantu expansion: insights from human paternal lineages. Mol Biol Evol. 2009;26:1581-1589.
[Google Scholar]
[CrossRef]
|
| 69. |
Verdu P, Becker NSA, Froment A, Georges M, Grugni V, Quintana-Murci L, et al. Sociocultural behavior, sex-biased admixture, and effective population sizes in Central African Pygmies and non-Pygmies. Mol Biol Evol. 2013;30:918-937.
[Google Scholar]
[CrossRef]
|
| 70. |
de Filippo C, Barbieri C, Whitten M, Mpoloka SW, Gunnarsdóttir ED, Bostoen K, et al. Y-chromosomal variation in sub-Saharan Africa: insights into the history of Niger-Congo groups. Mol Biol Evol. 2011;28:1255-1269.
[Google Scholar]
[CrossRef]
|
| 71. |
Anagnostou P, Battaggia C, Capocasa M, Boschi I, Brisighelli F, Batini C, et al. Reevaluating a model of gender-biased gene flow among Sub-Saharan Hunter-gatherers and farmers. Hum Biol. 2013;85:597-606.
[Google Scholar]
[CrossRef]
|
| 72. |
Verdu P, Austerlitz F. Post marital residence behaviours shape genetic variation in hunter-gatherer and agricultural populations from Central Africa. Hunter Gatherer Research. 2015;1(1):107-124.
[Google Scholar]
[CrossRef]
|
| 73. |
Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, et al. Genetic structure of human populations. Science. 2002;298:2381-2385.
[Google Scholar]
[CrossRef]
|
| 74. |
Patin E, Lopez M, Grollemund R, Verdu P, Harmant C, Quach H, et al. Dispersals and genetic adaptation of Bantu-speaking populations in Africa and North America. Science. 2017;356:543-546.
[Google Scholar]
[CrossRef]
|
| 75. |
Henn BM, Gignoux CR, Jobin M, Granka JM, Macpherson JM, Kidd JM, et al. Hunter-gatherer genomic diversity suggests a southern African origin for modern humans. Proc Natl Acad Sci USA. 2011;108:5154-5162.
[Google Scholar]
[CrossRef]
|
| 76. |
Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, et al. Ancient admixture in human history. Genetics. 2012;192:1065-1093.
[Google Scholar]
[CrossRef]
|
| 77. |
Lawson DJ, Hellenthal G, Myers S, Falush D. Inference of population structure using dense haplotype data. PLoS Genet. 2012;8:e1002453.
[Google Scholar]
[CrossRef]
|
| 78. |
Maples BK, Gravel S, Kenny EE, Bustamante CD. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am J Hum Genet. 2013;93:278-288.
[Google Scholar]
[CrossRef]
|
| 79. |
Goldberg A, Rosenberg NA. Beyond 2/3 and 1/3: The Complex Signatures of Sex-Biased Admixture on the X Chromosome. Genetics. 2015;201:263-279.
[Google Scholar]
[CrossRef]
|
| 80. |
Schiffels S, Durbin R. Inferring human population size and separation history from multiple genome sequences. Nat Genet. 2014;46:919-925.
[Google Scholar]
[CrossRef]
|
| 81. |
Shriver MD, Kennedy GC, Parra EJ, Lawson HA, Sonpar V, Huang J, et al. The genomic distribution of population substructure in four populations using 8,525 autosomal SNPs. Hum Genomics. 2004;1:274-286.
[Google Scholar]
[CrossRef]
|
| 82. |
Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913-918.
[Google Scholar]
[CrossRef]
|
| 83. |
Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72.
[Google Scholar]
[CrossRef]
|
| 84. |
Patin E, Quintana-Murci L. The demographic and adaptive history of central African hunter-gatherers and farmers. Curr Opin Genet Dev. 2018;53:90-97.
[Google Scholar]
[CrossRef]
|
| 85. |
Jarvis JP, Scheinfeldt LB, Soi S, Lambert C, Omberg L, Ferwerda B, et al. Patterns of ancestry, signatures of natural selection, and genetic association with stature in Western African pygmies. PLoS Genet. 2012;8:e1002641.
[Google Scholar]
[CrossRef]
|
| 86. |
Perry GH, Dominy NJ. Evolution of the human pygmy phenotype. Trends Ecol Evol. 2009;24:218-225.
[Google Scholar]
[CrossRef]
|
| 87. |
Hsieh P, Woerner AE, Wall JD, Lachance J, Tishkoff SA, Gutenkunst RN, et al. Model-based analyses of whole-genome data reveal a complex evolutionary history involving archaic introgression in Central African Pygmies. Genome Res. 2016;26:291-300.
[Google Scholar]
[CrossRef]
|
| 88. |
Lorente-Galdos B, Lao O, Serra-Vidal G, Santpere G, Kuderna LFK, Arauna LR, et al. Whole-genome sequence analysis of a Pan African set of samples reveals archaic gene flow from an extinct basal population of modern humans into sub-Saharan populations. Genome Biol. 2019;20:1-15.
[Google Scholar]
[CrossRef]
|
| 89. |
Goldstein DB, Chikhi L. Human migrations and population structure: what we know and why it matters. Annu Rev Genomics Hum Genet. 2002;3:129-152.
[Google Scholar]
[CrossRef]
|
| 90. |
Relethford JH. Genetics of modern human origins and diversity. Annu Rev Anthropol. 1998;27:1-23.
[Google Scholar]
[CrossRef]
|
| 91. |
Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M. Robust demographic inference from genomic and SNP data. PLoS Genet. 2013;9:e1003905.
[Google Scholar]
[CrossRef]
|
| 92. |
Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. Inferring the Joint Demographic History of Multiple Populations from Multidimensional SNP Frequency Data. PLoS Genet. 2009;5:e1000695.
[Google Scholar]
[CrossRef]
|
| 93. |
Jouganous J, Long W, Gravel S. Inferring the Joint Demographic History of Multiple Populations: Beyond the Diffusion Approximation. Genetics. 2017;206:1549-1567.
[Google Scholar]
[CrossRef]
|
| 94. |
Steinrücken M, Kamm J, Spence JP, Song YS. Inference of complex population histories using whole-genome sequences from multiple populations. Proc Natl Acad Sci USA. 2019;116:17115-17120.
[Google Scholar]
[CrossRef]
|
| 95. |
Arredondo A, Mourato B, Nguyen K, Boitard S, Rodríguez W, Noûs C, et al. Inferring number of populations and changes in connectivity under the n-island model. Heredity. 2021;126:896-912.
[Google Scholar]
[CrossRef]
|
| 96. |
Chikhi L, Rodríguez W, Grusea S, Santos P, Boitard S, Mazet O. The IICR (inverse instantaneous coalescence rate) as a summary of genomic diversity: insights into demographic inference and model choice. Heredity. 2018;120:13-24.
[Google Scholar]
[CrossRef]
|
| 97. |
Wang K, Mathieson I, O’Connell J, Schiffels S. Tracking human population structure through time from whole genome sequences. PLoS Genet. 2020;16:e1008552.
[Google Scholar]
[CrossRef]
|
| 98. |
Beaumont MA, Zhang W, Balding DJ. Approximate Bayesian computation in population genetics. Genetics. 2002;162:2025-2035.
[Google Scholar]
[CrossRef]
|
| 99. |
Tavaré S, Balding DJ, Griffiths RC, Donnelly P. Inferring coalescence times from DNA sequence data. Genetics. 1997;145:505-518.
[Google Scholar]
[CrossRef]
|
| 100. |
Wang K, Goldstein S, Bleasdale M, Clist B, Bostoen K, Bakwa-Lufu P, et al. Ancient genomes reveal complex patterns of population movement, interaction, and replacement in sub-Saharan Africa. Sci Adv. 2020;6:eaaz0183.
[Google Scholar]
[CrossRef]
|
| 101. |
Prendergast ME, Lipson M, Sawchuk EA, Olalde I, Ogola CA, Rohland N, et al. Ancient DNA reveals a multistep spread of the first herders into sub-Saharan Africa. Science. 2019;365.
[Google Scholar]
[CrossRef]
|
| 102. |
Durvasula A, Sankararaman S. Recovering signals of ghost archaic introgression in African populations. Sci Adv. 2020;6:eaax5097.
[Google Scholar]
[CrossRef]
|
| 103. |
Hsieh P, Veeramah KR, Lachance J, Tishkoff SA, Wall JD, Hammer MF, et al. Whole-genome sequence analyses of Western Central African Pygmy hunter-gatherers reveal a complex demographic history and identify candidate genes under positive natural selection. Genome Res. 2016;26:279-290.
[Google Scholar]
[CrossRef]
|
| 104. |
Lachance J, Vernot B, Elbers CC, Ferwerda B, Froment A, Bodo J-M, et al. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell. 2012;150:457-469.
[Google Scholar]
[CrossRef]
|
| 105. |
Ragsdale AP, Gravel S. Models of archaic admixture and recent history from two-locus statistics. PLoS Genet. 2019;15:e1008204.
[Google Scholar]
[CrossRef]
|
| 106. |
Fortes-Lima C, Schlebusch C. Closing the Gaps in Genomic Research. Trends Genet. 2021;37:104-106.
[Google Scholar]
[CrossRef]
|
| 107. |
Hammer MF, Woerner AE, Mendez FL, Watkins JC, Wall JD. Genetic evidence for archaic admixture in Africa. Proc Natl Acad Sci USA. 2011;108:15123-15128.
[Google Scholar]
[CrossRef]
|
| 108. |
Hey J, Chung Y, Sethuraman A, Lachance J, Tishkoff S, Sousa VC, et al. Phylogeny Estimation by Integration over Isolation with Migration Models. Mol Biol Evol. 2018;35:2805-2818.
[Google Scholar]
[CrossRef]
|
| 109. |
Quintana-Murci L. Human Immunology through the Lens of Evolutionary Genetics. Cell. 2019;177:184-199.
[Google Scholar]
[CrossRef]
|
| 110. |
Fagny M, Patin E, MacIsaac JL, Rotival M, Flutre T, Jones MJ, et al. The epigenomic landscape of African rainforest hunter-gatherers and farmers. Nat Commun. 2015;6:10047.
[Google Scholar]
[CrossRef]
|
| 111. |
Morton ER, Lynch J, Froment A, Lafosse S, Heyer E, Przeworski M, et al. Variation in Rural African Gut Microbiota Is Strongly Correlated with Colonization by Entamoeba and Subsistence. PLoS Genet. 2015;11:e1005658.
[Google Scholar]
[CrossRef]
|
| 112. |
Rubel MA, Abbas A, Taylor LJ, Connell A, Tanes C, Bittinger K, et al. Lifestyle and the presence of helminths is associated with gut microbiome composition in Cameroonians. Genome Biol. 2020;21:122.
[Google Scholar]
[CrossRef]
|
| 113. |
Gomez A, Petrzelkova KJ, Burns MB, Yeoman CJ, Amato KR, Vlckova K, et al. Gut Microbiome of Coexisting BaAka Pygmies and Bantu Reflects Gradients of Traditional Subsistence Patterns. Cell Rep. 2016;14:2142-2153.
[Google Scholar]
[CrossRef]
|
| 114. |
Wolfe ND, Dunavan CP, Diamond J. Origins of major human infectious diseases. Nature. 2007;447:279-283.
[Google Scholar]
[CrossRef]
|
| 115. |
Pagani L, Lawson DJ, Jagoda E, Mörseburg A, Eriksson A, Mitt M, et al. Genomic analyses inform on migration events during the peopling of Eurasia. Nature. 2016;538:238-242.
[Google Scholar]
[CrossRef]
|
| 116. |
Meyer M, Kircher M, Gansauge M-T, Li H, Racimo F, Mallick S, et al. A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012;338:222-226.
[Google Scholar]
[CrossRef]
|
| 117. |
Felsenstein J. PHYLIP (phylogeny inference package) version 3.695. Washington, USA: Distributed by the Author Department of Genome Sciences, University of Washington; 2013.
[Google Scholar]
|
| 118. |
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 2018;35:1547-1549.
[Google Scholar]
[CrossRef]
|


 ,
Cesar Fortes-Lima
1,†
,
Cesar Fortes-Lima
1,†