Introduction
Writing and commenting on one of the many seminal works Luigi Luca Cavalli-Sforza had conducted during his career is a tall order indeed. As one of the most influential scientists in the field of population genetics, he addressed such different aspects of human biology - from the genetic variability of human populations to its medical implications, from cultural anthropology and archaeology to linguistics - that talking about his scientific legacy with sufficient thoroughness sounds impossible. Indeed, as the first results of human genetic variability came out, he was a visionary who understood that the study of human evolutionary history was no longer just the remit of archaeologists and historians, thus turning human population genetics into a multidisciplinary field, with genes, language, pottery, and mathematical models sketching together our past.
The very same idea of shared knowledge drove the outset of the Human Genome Diversity Project (HGDP), the most complete and diverse worldwide human DNA collection, of which Cavalli-Sforza was an enthusiastic promoter. In this commentary, we will deal with the HGDP collection, its early days and groundbreaking results, as well as its controversies, starting from a paper published in 2005 by Cavalli-Sforza himself entitled “The Human Genome Diversity Project: past, present and future” [1]. Almost two decades have passed since then, during which the evolution of sequencing technologies has been recasting the human genetics field by allowing the production and comparison of genetic data from different human populations at an unparalleled scale. At the same time, the emergence and establishment of ancient DNA research have opened an unprecedented window into the major demographic events from our past. Following such scientific turmoils, the HGDP collection still remains a fruitful resource for the study of human variation, thanks to the high diversity of human ancestries enclosed in its populations.
The HGDP - Rationale and History
The HGDP, a collaborative endeavour aiming at exploring worldwide human genetic diversity, was born in 1991, pushed by the urge of preserving the record of our genetic heritage hidden, and back then still largely unexplored, within human genomes [2]. Until that time, the general lack of coordination among different studies led to the frustrating situation where samples were tested for different sets of genetic markers by different laboratories, thus preventing the possibility of comparing them to each other - the “empty matrix” problem [3]. At the same time, the progressing globalization was reaching even those populations which, due to geographical, linguistic or cultural barriers, had remained isolated for a long time, thus now “in danger” of losing their unique genetic makeup [4]. About that, Cavalli-Sforza and colleagues wrote: “It would be tragically ironic if, during the same decade that biological tools for understanding our species were created (the technical advancement of genotyping methods provided by the HGP – ndr), major opportunities for applying them were squandered” [2]. To overcome these issues, in the very same paper, they called for a project which would have: (i) collected human DNA from isolated and widely scattered human populations; (ii) stored renewable biological samples and their DNA and (iii) made both the DNA and the genotypes available to scientists. In this way, they launched the creation of a DNA repository and a database open to all researchers, which would be later called the Human Genome Diversity Project [3].
Despite the scientific novelty and soundness of the project, as well as its alluring goal of “understanding how and when patterns of diversity were formed” [1], the HGDP struggled for recognition, stuck in a planning phase for almost a decade, due to both political, economical and ethical issues. Whereas the purpose of HGDP of genotyping sets of markers from different populations would have required only a fraction of the time and money necessary for the complete sequencing of the human genome (HGP), lack of funding was hampering the implementation of the project. Moreover, although the HGDP took into great consideration the importance of ethical issues since the initial phases [5], its intent of exploring the genetic richness of the entire human species inevitably and directly touches our intimate human nature and beliefs, which may be different depending on our scientific background or culture. It is therefore quite natural - and advisable - that concerns moved by various sensitivities and expertise arose from different scientific disciplines, as well as from society. The main issues raised by anthropologists, ethicists and some of the Indigenous communities regarded the fear of drifting towards the economical exploitation of the communities’ DNA (biopiracy or biocolonialism) or the possibility that the HGDP results would feed “scientific racism”. Moreover, the very same “open science” idea at the root of HGDP - making biological samples, DNA and the resulting data available to scientists - implied that such materials would have been analysed by an unknown number of researchers, for an indefinite time and a broad range of scientific goals, thus raising additional issues about informed consent and secondary uses [3].
The HGDP recognized the importance of the ethical, legal and social aspects involving DNA collection and defined its internal protocols after years of multidisciplinary discussion about the optimal procedures to be used for carrying on such a sensitive task of collecting and analysing humanity’s genetic diversity [5]. Such guidelines have then been reviewed by the US National Academy of Sciences National Research Council [6] and constantly supervised by the NIH Institute of General Medical Sciences. HGDP always avoided commercial interests by granting access to academic research only and research performed on its data contributed to demonstrating that there is no scientific basis for racism [7–9]. Nevertheless, some of the past ethical issues - involving difficult scientific and social questions about the correct administration of informed consent and secondary uses - were not completely overcome and are still relevant today for human genomic research when accessible population DNA resources are designed. Moreover, despite the efforts Cavalli-Sforza and collaborators put into drawing guidelines for respectful sample collection strategies [5], in some cases, the lack of meaningful engagement with Indigenous communities led to misunderstandings and mistrust, thus hampering future scientific endeavours and contributing to the underrepresentation of Indigenous populations in genomic studies [10].
For what concerns the biological material to be collected, the HGDP organizers decided to rely on lymphoblastoid cell lines (LCLs) instead of just taking extracted DNA samples, thus guaranteeing an indefinite supply of DNA. The collection is stored at the Center for the Study of Human Polymorphism (CEPH) at the Foundation Jean Dausset and, for this reason, the population database has been referred to as HGDP-CEPH.
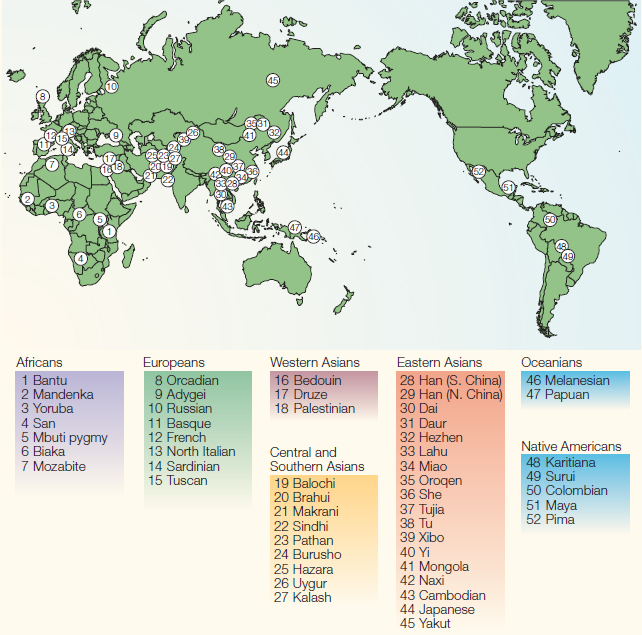
Starting from 2002, the DNA from 1064 LCLs, as well as the information on sex, population, and geographic origin of the individuals, were made available to researchers agreeing to deposit their results to a central database [11]. All five continents were represented in the HGDP-CEPH, with individuals coming from 51 anthropologically relevant populations from cultural, linguistical or historical points of view (Figure 1). For instance, the collection process focuses mainly on those populations predating the voyages of discovery started in the fifteenth and sixteenth centuries and, while being extremely informative from a scientific point of view, this criterion was also harshly criticised by some of the Indigenous groups who felt they were being considered “living fossils”. This sampling strategy means that the HGDP-CEPH is not a random sample of the worldwide populations [8], with some areas (e.g., China and Pakistan) being more represented than others (Africa, America and Oceania).
However, twenty years later, the HGDP-CEPH remains “the most complete worldwide human DNA collection that is available to not-for-profit researchers” [1], thanks to its anthropological-driven study design. From the very first large-scale population genetics study in 2002 [7] to its whole-genome sequencing sequel [12], it has been feeding our knowledge of human genetic variation in such diverse fields as human evolution, anthropology, forensic genetics, as well as medical studies.
The HGDP - Twenty Years of Human Genetic Variation Studies
Population genetic variability across time and space
The contribution that the HGDP-CEPH collection, together with other mostly coeval population-based sampling efforts [13–15], gave to the understanding of the human genetic makeup is priceless. Several reanalyses of the HGDP-CEPH samples with different genetic assays followed one another, thus consolidating and introducing some fundamental knowledge about human genome diversity, such as our African origins supported by serial founder effects and the mechanisms of selection and drift shaping the patterns of worldwide genetic variability, as well as refining and expanding the proposed models of historical human migration [7–9,12,16–26].
In the first years after the HGDP-CEPH setup, three papers analyzing 377 microsatellites and half a million single nucleotide polymorphisms (SNPs) on HGDP-CEPH samples were published [7–9], the latter two paving the way to SNP-based studies of human genetic variation. They confirmed that the larger portion of human genetic diversity is explained by differences between individuals, instead of between-populations or between-groups variability. This fact strongly highlighted the overall similarity of human populations, as expected from the recent history of our species, which is characterized by extensive gene flow. Nevertheless, they also described the substantial genetic structure of worldwide human populations which roughly correspond to the five continents. This result was achieved thanks to the high number of individuals and genetic markers typed, which allowed us to appreciate the subtle allele frequency differences across the loci responsible for such structure. These two findings - which may appear contradictory at first and have been indeed perceived as such by the general public with detrimental consequences on “race” debates - can be explained by the fact that most alleles are widespread, with their frequency differences among populations being distributed in gradients rather than locking this or that populations in their own impassable borders.
Around twenty years later, in 2020, almost the entire HGDP-CEPH collection (929 samples) was sequenced to an average coverage of 35X in a paper published in Science, with whole-genome sequences being freely available to all researchers [12]. While large-scale population sequencing projects had been extensively conducted until then, they mainly focused on metropolitan individuals employing usually low-coverage sequencing technologies [27–29]. On the other hand, when instead geographically diverse populations were addressed, just a few individuals per population have been sequenced [21,30]. The results of Bergström and colleagues strongly demonstrated that anthropologically-driven genomic studies provide a deeper understanding of human genetic variation than other study designs, through, for instance, an increased variant discovery power. Indeed, the sequenced HGDP-CEPH panel harboured nearly as many genetic variants as the 1000 Genomes Project (1KGP, 67.3 million and 84.7 million SNPs, respectively), while having half of their samples. If from one side, this could be explained by the increased coverage of the Bergström study, an important role is played by the increased genetic diversity of human populations covered by the HGDP-CEPH collection. Interestingly, the HGDP-CEPH contained a considerable number of alternative alleles that even though common or even high-frequency in some populations, were not present in the 1KGP: ~1 million variants at ≥20%, ~100,000 variants at ≥50%, and around 1000 variants fixed at 100% frequency in at least one population sample, with geographical areas such as Africa, Oceania and America harbouring the highest fractions. However, no private variants that are fixed in a continent or major geographical area (i.e., present in 100% of genomes) have been found, thus supporting the knowledge that the majority of common genetic variation is shared across the globe.
Many other studies investigated instead human groups included in the HGDP characterized by geographical isolation or peculiar linguistic and cultural traits in order to retrace their origin. For instance, the distinct genetic makeups of Sardinians and Basques, with respect to other European populations, have always cast doubts on their origins, which turned out to be mostly Neolithic for the former and the result of genetic continuity since the Iron Age for the latter [31–35].
It is interesting to note that since the HGDP conception, about thirty years ago, major breakthroughs have shaken the foundation of human genetics, from the introduction of next generation sequencing technologies to the recent complete human genome assembly [36]. The population genetic field was shaken too by another equally stunning innovation, ancient DNA. The discoveries made thanks to the DNA isolated from ancient human and hominin fossils are rewriting human history, some examples being the admixture between early humans and Neanderthals and the existence of Denisova, an archaic human species that has yet to be physically characterized [37,38]. However, exploring the genetic variations of ancient and archaic humans in order to reconstruct their population dynamics required modern human genetic data as a reference. The highly divergent ancestries enclosed within the HGDP-CEPH individuals seemed the best candidates to put the sometimes outsider ancient variability into the context of worldwide modern human genetic variations. For example, 5 HGDP-CEPH individuals (Han Chinese, Yoruba, San, Papuan and French) were specifically sequenced in order to answer the long-standing questions about our ancestors’ relationships with Neanderthals, when the first draft of the Neanderthal genome became available [37]. In the same year, the same individuals were then compared with the Denisovan genome and one of them - the Papuan - was decisive to highlight gene flow from the Denisovans [38]. While sequencing the entire HGDP-CEPH panel was unaffordable, commercial SNP array platforms, originally designed for genome-wide association studies on mostly European populations, were known to suffer from ascertainment biases, thus significantly affecting population genetic inferences [39]. In order to overcome this issue, Patterson and colleagues assembled and validated an array consisting of 600,000 SNPs selected from 11 different modern human populations, as well as archaic hominids and chimp [17], which was called the Human Origin Array. The HGDP-CEPH panel was chosen to validate the array, the final genotyping data were made freely available to the public and, since then, the HGDP-CEPH worldwide populations genotyped with Human Origin Array represented a so far irreplaceable reference for ancient DNA data. In the same seminal paper, they presented also a new mathematical framework, the so-called f-statistics, for formally modelling population mixtures through “admixture graphs” (i.e., phylogenetic tree-based models) which are fitted to genetic data. Results from f-statistics-based methods (e.g., F2, F3, F4 for 2, 3 and 4 populations, respectively) hold rigorously if an outgroup (i.e., a population having the same genetic relationship with populations A, B and C, in case of an F4) is identified and the analysis is restricted to SNPs which are polymorphic in the outgroup. These methods have gradually become one of the gold standards in admixture inference analyses and, when Non-African populations are investigated, the Mbuti central African rainforest hunter-gatherers from the HGDP-CEPH panel played a crucial role as the ideal unbiased outgroup for all Non-African populations, given the negligible fraction of Eurasian admixture detected in this population.
Genetic variability and medical implications
While one of the stated goals of the HGDP was to foster medical research, the lack of phenotypic information and the discrete number of individuals from each location hindered its direct application in medical research. Indeed, the chosen populations were more useful for framing worldwide human genetic diversity than for providing a good reference for the metropolitan populations that were the most studied in medical genetics. When the sequencing of the human genome and the early exploration of genetic diversity spurred the big wave of Genome Wide Association Studies (GWAS) that began in 2005 [40], sample sizes for controls soon outgrew the HGDP-CEPH numbers by orders of magnitude [41], making the HGDP direct impact on medical genetics negligible, especially when compared to other projects like HapMap [13].
However, the undeniable importance of the HGDP in population genetics also indirectly impacted the medical field. The deluge of studies exploring human genetic diversity provided a valuable framework for the design and interpretation of medical studies. Indeed, a deep knowledge of the genetic structure and variability within human populations is extremely relevant in order to better identify the genetic architecture of complex traits or diseases. GWAS rely heavily on this knowledge to control biases and avoid spurious signals due to population stratification and while the HGDP individuals saw little use as controls, their diversity made them an invaluable compass to use for mapping the stratification in the metropolitan populations commonly examined in GWAS. Another example was in the development of genetic imputation, where the HGDP samples were chosen for the peculiar genetic makeup of some of their populations to test the limits of the available imputation panels [42].
The importance of population structure continues to be relevant even nowadays: from the thousands of GWAS performed so far [43], it becomes clear that the polygenic contribution to complex traits and diseases defined by the Polygenic Risk Scores (PRS) are quite variable among populations of different ancestries [44], thus limiting the clinical applicability and transferability of the PRS. Indeed, the main ingredients of PRS are the GWAS summary statistics, which have been mainly built on samples of European origins [45]. For this reason, the traditional lack of diversity in genomic studies, which is currently denying the potential benefits of genomic research to underrepresented populations, has been addressed in the last years by many initiatives working against the Eurocentric biases of genomics [46].
Advances in sequencing technologies have highlighted the fundamental role played by the reference sequence, both in terms of challenging unresolved regions and the need to represent the full diversity of human populations. In this context, two scientific endeavours are working towards addressing these issues: the Telomere-to-Telomere Consortium and the Human Pangenome Reference Consortium [47,48]. Indeed, a new update of the human genome sequence was released in April 2022 [47], including more than 200 Mb not covered by the previous versions and containing more than 1900 genes, now resolved mainly by long-read third generation sequencing technologies which allow a more accurate reconstruction of the haplotype structure across the genome, as well as the improvement of short-read mapping and variant calling across populations. On the other hand, the Human Pangenome Reference Consortium is currently working towards a more precise, inclusive - in terms of human genomic diversity - and graph-based reference sequence [48], with a goal of assembling at least 350 reference quality haplotype-phased human genomes. The two initiatives are pursuing the common aim of improving the human reference genome together, with important consequences for variant discovery across populations in the next years. Hopefully, stored samples of HGDP populations could still be of great help if used also for this purpose, as anticipated by the recent whole-genome sequencing of the HGDP-CEPH samples revealing a much higher proportion of common and rare variability, compared with the 1KGP [12].
Other genomic projects
The HGDP is just one of many projects that tackled human genetics in the last twenty years (see Table 1). After the Human Genome Project (HGP) yielded the first version of the human genome, that characterizes us as a species, there was naturally a need to discover the human genetic variation, where everything that made us individuals was to be found. Indeed, in the 0.1% of the genome that varies between individuals, there was a wealth of information, both for population and medical genetics. Thus, the knowledge and the technological and methodological advances yielded by the HGP were followed closely by a stream of other projects aimed at collecting more genetic diversity, that in turn yielded more knowledge and new technologies, enabling further and more ambitious research.
One of the first was the HapMap project, starting right after the completion of the HGDP in 2002. Similar to HGDP, its broad objective was to explore human genetic diversity, but with different priorities and a different strategy. It prioritized enabling medical studies by discovering common human variations and focusing on the patterns of linkage disequilibrium to find a collection of tag SNPs that could be used to detect genotype-phenotype associations without having to resort to prohibitive sequencing. As a consequence, it collected larger samples from metropolitan populations that were commonly studied in medical practice instead of genetically isolated or peculiar populations as in HGDP. This approach proved to be more fruitful for medical genetics (and less ethically questionable), with knowledge of common variation and linkage structure allowing the development of cheap genotyping microarrays that ushered in the era of GWAS. HapMap was followed by the 1000 Genomes Project [27,49], which expanded the sampled populations and used sequencing, producing whole genome sequencing data of 2504 individuals worldwide.
Despite their differences, the HGDP, HapMap and the 1KGP also had many similarities: they collected individuals following population criteria, used immortalized cell lines to guarantee the possibility of further analyses with newer technologies and made individual genetic data publicly available without restrictions.
These features set them apart from a generation of more recent efforts like ExAC [28] and gnomAD [29]. These are just the largest of a series of projects that gathered raw sequencing data from existing studies and merged them into extensive datasets with more than a hundred thousand individuals from all over the world. While this approach allowed the collection of an unprecedented amount of data on human variation, it had important implications. One was that the anthropologic characterization of the collected individuals was lacking, with only coarse-grained information about their geographical origin. Another was that individual genetic data was never made public. While the projects’ discoveries had a profound impact on genetics and the aggregated allele frequency database soon became an essential resource in medical genetics, the impact on population genetics was more modest. The two limitations we mentioned severely hampered both the direct discoveries and the reuse of these large datasets in new population studies. Another endeavour that followed a similar strategy was the Haplotype Reference Consortium (HRC), which gathered WGS data of tens of thousands in order to surpass the 1KGP as an imputation panel. Again, the individual data could not be released and the imputation could only be performed on the Consortium servers.
Yet another different type of initiative is represented by the population biobanks, which gather biological samples and phenotype data from individuals that are then followed for decades to observe health-related outcomes. Often, they are linked to the national health system, like the Estonian and UK Biobanks, established in 2000 and 2004, respectively. The biological samples allow subsequent studies to generate different types of molecular data. Indeed, now many biobanks offer a wealth of genetic and phenotypic data (on request) for scientific studies. Cavalli-Sforza was well aware of these projects [1] and hoped that they could contribute cell lines to the HGDP-CEPH collection. Unfortunately, this never happened.
All these are important efforts to discover and understand human genetic diversity and they had a scientific impact that cannot be overstated, both by the knowledge they yielded and by enabling further studies. However, even twenty years after its first results, the HGDP-CEPH remains the only collection of individuals that can represent the worldwide variability present in non-metropolitan populations with freely available individual genetic data. For this reason, the HGDP continues to play an essential role in population genetics and in framing the genetic diversity of the human species.
Beyond the HGDP - New Challenges and Outlooks for Human Genetic Variation Studies
In the last twenty years, the HGDP has shaped the development of the entire field of population genetics. Thanks to the availability of immortalized cell lines, the collection was reanalyzed multiple times with newer technologies, each time providing more data and new or more precise answers. This led to the many studies that focused on the collection itself, from the first study by Rosenberg and colleagues [7] to the recent whole-sequencing of the panel [12]. Moreover, countless other research efforts that focused on other modern populations or even ancient DNA have relied on the HGDP-CEPH samples as an essential reference for framing human genetic diversity. This is further testified by the widely used Human Origin Array [12,17], which owes its design to the HGDP-CEPH samples.
Despite its value to scientific research and the wishes of Cavalli-Sforza, the HGDP did not grow after the initial collection of samples and few if any cell lines were added in these two decades. Neither did a direct successor project appear, with comparable goals and scope. However, the HGDP-derived data was repeatedly merged with new population data from more focused studies [50–52]. In particular, this is somewhat straightforward when using sequencing data from both the HGDP [12] and other genomic projects for which individual genotypes are available (Table 1). A cautionary note regards the possible technical biases coming from different sequencing platforms and methodologies which may result in the need of reprocessing the data starting from raw reads [53].
A relevant challenge for the future of the HGDP, but also possibly of HapMap and the 1KGP, is the growing attention to data protection, which has grown substantially during the life of these projects. Public and legal debate over privacy issues in the last years was condensed for instance in the General Data Protection Regulation (GDPR), with far-reaching consequences in the handling of genetic data. One of them is that, since a person must retain ownership and control over their personal data, it is becoming harder and harder for individual human genetic data to be published without restrictions. While the HGDP-derived datasets have been published again and again, to the point where it would be futile to try to regain control of the data, the CEPH foundation does not provide access to individual genetic data anymorei.
Among the discussed ethical issues raised initially around the HGDP project, including the little benefit for the individuals and populations sampled and the risk of discriminatory misuses of scientific facts and arguments for which the HGDP promoters cannot be responsible, the HGDP has anticipated the need of sharing data among researchers in order to accelerate scientific knowledge. To share genetic data in an ethical way that also complies with privacy regulations such as the GDPR, several initiatives moved recently in this direction such as the Global Alliance for Genomics and Health (GA4GH) with the goal to promote scientific research by favouring responsible sharing of clinical and genomic data. The GA4GH works to set up policy frameworks and technical standards that are both secure and interoperable. Wide support is essential when working on standards and frameworks that need to be adopted and deployed in order to make the most of this effort in data sharing that could be key to the future of genomic medicine and especially to making sure that everybody, regardless of ancestry, can benefit from it.
In conclusion, the HGDP has been a pioneering project characterized by interdisciplinary endeavours since its outset. As the first of its kind, working toward a comprehensive knowledge of human genetic variation, it has drawn during the years also serious criticisms. While some of them were never addressed by the HGDP itself, they fed a healthy debate on the ethical challenges of genetic research, giving birth to new questions, tackling different sensibilities, and yielding new answers. Later projects on genetic diversity benefited from such debates, thus finally advancing the original goal of the HGDP: adding one more “unique thread to the tapestry of our knowledge of humanity” [5].
Declarations
Competing Interests
The authors declare no competing interests.
Acknowledgement
This work was supported by TESEO (Achievements of Excellence in Medical Sciences Exploring the Omics). Department of Medical Sciences of the Italian Ministry for Education, University and Research (Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR)) under the program “Dipartimenti di Eccellenza 2018–2022”. Project no. D15D18000410001.


 ,
Giovanni Birolo
2
,
Giovanni Birolo
2